Bacterial Communities Associated with Atherosclerotic Plaques from Russian Individuals with Atherosclerosis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

INOCULUM DYNAMICS of Ralstonia Spp.: POTENTIAL SOURCES, PERSISTENCE in a LOCAL POPULATION and SELECTION of PHAGES to REDUCE BACTERIA SURVIVAL
JAQUELINE KIYOMI YAMADA INOCULUM DYNAMICS OF Ralstonia spp.: POTENTIAL SOURCES, PERSISTENCE IN A LOCAL POPULATION AND SELECTION OF PHAGES TO REDUCE BACTERIA SURVIVAL Thesis presented to the Plant Pathology Program of the Universidade Federal de Viçosa in partial fulfillment of the requirements for the degree of Doctor Scientiae. VIÇOSA MINAS GERAIS – BRASIL 2018 ) AGRADECIMENTOS Agradeço a Deus, pela vida, por estar presente em todos os momentos. Agradeço aos meus pais, Jorge e Helena, exemplos de honestidade e humildade, todas as minhas conquistas são fruto do sacrifício deles. À minha irmã, Michelle, por todo apoio. Obrigada Mi! Agradeço ao Filipe Constantino Borel, pelo companheirismo e carinho, fundamental para a conclusão dessa etapa da minha vida. Agradeço à Nina e Júlio Borel, pais de Filipe, por todo apoio. Agradeço à Universidade Federal de Viçosa, ao Departamento de Fitopatologia e à FAPEMIG pela oportunidade e pelo financiamento desse trabalho. Agradeço ao Professor Eduardo Seite Gomide Mizubuti pela oportunidade, paciência e conselhos. Agradeço ao Doutor Carlos Alberto Lopes, pela contribuição para o presente trabalho e pela amizade. Agradeço ao Professor José Rogério e ao Professor Francisco Murilo Zerbini por terem disponibilizados os laboratórios para realização desse trabalho. Agradeço ao Professor Sérgio Oliveira de Paula, em especial ao Roberto de Sousa Dias e ao Vinicius Duarte pela colaboração e pela amizade. Agradeço à Thaís Ribeiro Santiago e à Camila Geovanna Ferro, pelas sugestões e pela amizade. Agradeço a todos que colaboraram com as amostras de solo e água para este trabalho: Paulo E. F. de Macedo, Amanda Guedes, Carla Santin, Leandro H. Yamada, Filipe C. -

Bacterial Diseases of Bananas and Enset: Current State of Knowledge and Integrated Approaches Toward Sustainable Management G
Bacterial Diseases of Bananas and Enset: Current State of Knowledge and Integrated Approaches Toward Sustainable Management G. Blomme, M. Dita, K. S. Jacobsen, L. P. Vicente, A. Molina, W. Ocimati, Stéphane Poussier, Philippe Prior To cite this version: G. Blomme, M. Dita, K. S. Jacobsen, L. P. Vicente, A. Molina, et al.. Bacterial Diseases of Bananas and Enset: Current State of Knowledge and Integrated Approaches Toward Sustainable Management. Frontiers in Plant Science, Frontiers, 2017, 8, pp.1-25. 10.3389/fpls.2017.01290. hal-01608050 HAL Id: hal-01608050 https://hal.archives-ouvertes.fr/hal-01608050 Submitted on 28 Aug 2019 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Distributed under a Creative Commons Attribution| 4.0 International License fpls-08-01290 July 22, 2017 Time: 11:6 # 1 REVIEW published: 20 July 2017 doi: 10.3389/fpls.2017.01290 Bacterial Diseases of Bananas and Enset: Current State of Knowledge and Integrated Approaches Toward Sustainable Management Guy Blomme1*, Miguel Dita2, Kim Sarah Jacobsen3, Luis Pérez Vicente4, Agustin -

Whole Genome Characterization of Strains Belonging to the Ralstonia
Eur J Plant Pathol https://doi.org/10.1007/s10658-020-02190-8 Whole genome characterization of strains belonging to the Ralstonia solanacearum species complex and in silico analysis of TaqMan assays for detection in this heterogenous species complex Viola Kurm & Ilse Houwers & Claudia E. Coipan & Peter Bonants & Cees Waalwijk & Theo van der Lee & Balázs Brankovics & Jan van der Wolf Accepted: 17 December 2020 # The Author(s) 2021 Abstract Identification and classification of members of that the increasing availability of whole genome sequences the Ralstonia solanacearum species complex (RSSC) is is not only useful for classification of strains, but also shows challenging due to the heterogeneity of this complex. Whole potential for selection and evaluation of clade specific genome sequence data of 225 strains were used to classify nucleic acid-based amplification methods within the RSSC. strains based on average nucleotide identity (ANI) and multilocus sequence analysis (MLSA). Based on the ANI Keywords MLSA . ANI . in-silico analysis . Ralstonia score (>95%), 191 out of 192(99.5%) RSSC strains could solanacearum species complex . Phylogenetic be grouped into the three species R. solanacearum, R. classification pseudosolanacearum,andR. syzygii, and into the four phylotypes within the RSSC (I,II, III, and IV). R. solanacearum phylotype II could be split in two groups Introduction (IIA and IIB), from which IIB clustered in three subgroups (IIBa, IIBb and IIBc). This division by ANI was in accor- Bacteria belonging to the Ralstonia solanacearum spe- dance with MLSA. The IIB subgroups found by ANI and cies complex (RSSC) are the causative agents of dis- MLSA also differed in the number of SNPs in the primer eases in plants of many different botanical families. -
![Pangenomic Type III Effector Database of the Plant Pathogenic [I]Ralstonia Spp.[I]](https://docslib.b-cdn.net/cover/1381/pangenomic-type-iii-effector-database-of-the-plant-pathogenic-i-ralstonia-spp-i-611381.webp)
Pangenomic Type III Effector Database of the Plant Pathogenic [I]Ralstonia Spp.[I]
A peer-reviewed version of this preprint was published in PeerJ on 6 August 2019. View the peer-reviewed version (peerj.com/articles/7346), which is the preferred citable publication unless you specifically need to cite this preprint. Sabbagh CRR, Carrere S, Lonjon F, Vailleau F, Macho AP, Genin S, Peeters N. 2019. Pangenomic type III effector database of the plant pathogenic Ralstonia spp. PeerJ 7:e7346 https://doi.org/10.7717/peerj.7346 Pangenomic type III effector database of the plant pathogenic Ralstonia spp. Cyrus Raja Rubenstein Sabbagh Equal first author, 1 , Sébastien Carrère Equal first author, 1 , Fabien Lonjon 2 , Fabienne Vailleau 1 , Alberto P Macho 3 , Stephane Genin 1 , Nemo Peeters Corresp. 1 1 LIPM, Université de Toulouse, INRA, CNRS, Castanet-tolosan, France 2 Department of Cell & Systems Biology, University of Toronto, Toronto, Canada 3 Shanghai center for plant stress biology, CAS Center for Excellence in Molecular Plant Sciences, Shanghai Institutes of Biological Sciences, Chinese Academy of Sciences, Shanghai, China Corresponding Author: Nemo Peeters Email address: [email protected] Background. The bacterial plant pathogenic Ralstonia species belong to the beta- proteobacteria order and are soil-borne pathogens causing the vascular bacterial wilt disease, affecting a wide range of plant hosts. These bacteria form a heterogeneous group considered as a “species complex”,” gathering three newly defined species. Like many other Gram negative plant pathogens, Ralstonia pathogenicity relies on a type III secretion system, enabling bacteria to secrete/inject a large repertoire of type III effectors into their plant host cells. T3Es are thought to participate in generating a favorable environment for the pathogen (countering plant immunity and modifying the host metabolism and physiology). -

Microbial Communities in Placentas from Term Normal Pregnancy Exhibit Spatially Variable Profiles Lindsay A
Washington University School of Medicine Digital Commons@Becker Open Access Publications 2017 Microbial communities in placentas from term normal pregnancy exhibit spatially variable profiles Lindsay A. Parnell Washington University School of Medicine in St. Louis Catherine M. Briggs Washington University School of Medicine in St. Louis Bin Cao Washington University School of Medicine in St. Louis Omar Delannoy-Bruno Washington University School of Medicine in St. Louis Andrew E. Schrieffer Washington University School of Medicine in St. Louis See next page for additional authors Follow this and additional works at: https://digitalcommons.wustl.edu/open_access_pubs Recommended Citation Parnell, Lindsay A.; Briggs, Catherine M.; Cao, Bin; Delannoy-Bruno, Omar; Schrieffer, Andrew E.; and Mysorekar, Indira U., ,"Microbial communities in placentas from term normal pregnancy exhibit spatially variable profiles." Scientific Reports.7,. (2017). https://digitalcommons.wustl.edu/open_access_pubs/6175 This Open Access Publication is brought to you for free and open access by Digital Commons@Becker. It has been accepted for inclusion in Open Access Publications by an authorized administrator of Digital Commons@Becker. For more information, please contact [email protected]. Authors Lindsay A. Parnell, Catherine M. Briggs, Bin Cao, Omar Delannoy-Bruno, Andrew E. Schrieffer, and Indira U. Mysorekar This open access publication is available at Digital Commons@Becker: https://digitalcommons.wustl.edu/open_access_pubs/6175 www.nature.com/scientificreports OPEN Microbial communities in placentas from term normal pregnancy exhibit spatially variable profles Received: 30 January 2017 Lindsay A. Parnell1, Catherine M. Briggs1, Bin Cao1, Omar Delannoy-Bruno1, Andrew E. Accepted: 24 August 2017 Schriefer2 & Indira U. Mysorekar 1,3 Published: xx xx xxxx The placenta is the principal organ nurturing the fetus during pregnancy and was traditionally considered to be sterile. -

A Genome-Wide Scan for Genes Under Balancing Selection in the Plant Pathogen Ralstonia Solanacearum José A
Castillo and Agathos BMC Evolutionary Biology (2019) 19:123 https://doi.org/10.1186/s12862-019-1456-6 RESEARCHARTICLE Open Access A genome-wide scan for genes under balancing selection in the plant pathogen Ralstonia solanacearum José A. Castillo1* and Spiros N. Agathos2 Abstract Background: Plant pathogens are under significant selective pressure by the plant host. Consequently, they are expected to have adapted to this condition or contribute to evading plant defenses. In order to acquire long-term fitness, plant bacterial pathogens are usually forced to maintain advantageous genetic diversity in populations. This strategy ensures that different alleles in the pathogen’s gene pool are maintained in a population at frequencies larger than expected under neutral evolution. This selective process, known as balancing selection, is the subject of this work in the context of a common bacterial phytopathogen. We performed a genome-wide scan of Ralstonia solanacearum species complex, an aggressive plant bacterial pathogen that shows broad host range and causes a devastating disease called ‘bacterial wilt’. Results: Using a sliding window approach, we analyzed 57 genomes from three phylotypes of the R. solanacearum species complex to detect signatures of balancing selection. A total of 161 windows showed extreme values in three summary statistics of population genetics: Tajima’sD,θw and Fu & Li’s D*. We discarded any confounding effects due to demographic events by means of coalescent simulations of genetic data. The prospective windows correspond to 78 genes with known function that map in any of the two main replicons (1.7% of total number of genes). The candidate genes under balancing selection are related to primary metabolism and other basal activities (51.3%) or directly associated to virulence (48.7%), the latter being involved in key functions targeted to dismantle plant defenses or to participate in critical stages in the pathogenic process. -

Cas Systems in the Ralstonia Solanacearum Species Complex
bs_bs_banner MOLECULAR PLANT PATHOLOGY (2019) 20(2), 223–239 DOI: 10.1111/mpp.12750 Characterization of CRISPR-Cas systems in the Ralstonia solanacearum species complex ANDRÉ DA SILVA XAVIER1, JULIANA CRISTINA FRALEON DE ALMEIDA1, ALESSANDRA GONÇALVES DE MELO2, GENEVIÈVE M. ROUSSEAU2,3, DENISE M. TREMBLAY2,3, RAFAEL REIS DE REZENDE1, SYLVAIN MOINEAU2,3 AND POLIANE ALFENAS-ZERBINI1* 1 Departamento de Microbiologia, Instituto de Biotecnologia Aplicada à Agropecuária (BIOAGRO), Universidade Federal de Viçosa, Viçosa, MG 36570-000, Brazil 2 Département de Biochimie, de Microbiologie, et de Bioinformatique, Faculté des Sciences et de Génie, Université Laval, Québec City, QC GIV0A6, Canada 3 Félix d’Hérelle Reference Center for Bacterial Viruses, and GREB, Faculté de Médecine Dentaire, Université Laval, Québec City, QC GIV0A6, Canada SUMMARY INTRODUCTION Clustered regularly interspaced short palindromic repeats The Gram-negative plant-pathogenic bacteria Ralstonia spp. be- (CRISPRs) are composed of an array of short DNA repeat se- long to a species complex, the Ralstonia solanacearum species quences separated by unique spacer sequences that are complex (RSSC), which is recognized as a group of considerable flanked by associated (Cas) genes. CRISPR-Cas systems are genetic diversity encompassing phenotypically diverse strains found in the genomes of several microbes and can act as an that can be subdivided into four phylotypes (Allen et al., 2005; adaptive immune mechanism against invading foreign nucleic Prior and Fegan, 2005). Phylotypes I, II and III contain strains acids, such as phage genomes. Here, we studied the CRISPR- predominantly from Asia, America and Africa and surrounding is- Cas systems in plant-pathogenic bacteria of the Ralstonia sola- lands, respectively, whereas phylotype IV is comprised of strains nacearum species complex (RSSC). -

Sparus Aurata) and Sea Bass (Dicentrarchus Labrax)
Gut bacterial communities in geographically distant populations of farmed sea bream (Sparus aurata) and sea bass (Dicentrarchus labrax) Eleni Nikouli1, Alexandra Meziti1, Efthimia Antonopoulou2, Eleni Mente1, Konstantinos Ar. Kormas1* 1 Department of Ichthyology and Aquatic Environment, School of Agricultural Sciences, University of Thessaly, 384 46 Volos, Greece 2 Laboratory of Animal Physiology, Department of Zoology, School of Biology, Aristotle University of Thessaloniki, 541 24 Thessaloniki, Greece * Corresponding author; Tel.: +30-242-109-3082, Fax: +30-242109-3157, E-mail: [email protected], [email protected] Supplementary material 1 Table S1. Body weight of the Sparus aurata and Dicentrarchus labrax individuals used in this study. Chania Chios Igoumenitsa Yaltra Atalanti Sample Body weight S. aurata D. labrax S. aurata D. labrax S. aurata D. labrax S. aurata D. labrax S. aurata D. labrax (g) 1 359 378 558 420 433 448 481 346 260 785 2 355 294 579 442 493 556 516 397 240 340 3 376 275 468 554 450 464 540 415 440 500 4 392 395 530 460 440 483 492 493 365 860 5 420 362 483 479 542 492 406 995 6 521 505 506 461 Mean 380.40 340.80 523.17 476.67 471.60 487.75 504.50 419.67 326.25 696.00 SEs 11.89 23.76 17.36 19.56 20.46 23.85 8.68 21.00 46.79 120.29 2 Table S2. Ingredients of the diets used at the time of sampling. Ingredient Sparus aurata Dicentrarchus labrax (6 mm; 350-450 g)** (6 mm; 450-800 g)** Crude proteins (%) 42 – 44 37 – 39 Crude lipids (%) 19 – 21 20 – 22 Nitrogen free extract (NFE) (%) 20 – 26 19 – 25 Crude cellulose (%) 1 – 3 2 – 4 Ash (%) 5.8 – 7.8 6.2 – 8.2 Total P (%) 0.7 – 0.9 0.8 – 1.0 Gross energy (MJ/Kg) 21.5 – 23.5 20.6 – 22.6 Classical digestible energy* (MJ/Kg) 19.5 18.9 Added vitamin D3 (I.U./Kg) 500 500 Added vitamin E (I.U./Kg) 180 100 Added vitamin C (I.U./Kg) 250 100 Feeding rate (%), i.e. -

Community Structure of Soil Bacteria in a Tropical Rainforest Several Years After Fire
Microbes Environ. Vol. 23, No. 1, 49–56, 2008 http://wwwsoc.nii.ac.jp/jsme2/ doi:10.1264/jsme2.23.49 Community Structure of Soil Bacteria in a Tropical Rainforest Several Years After Fire SHIGETO OTSUKA1*, IMADE SUDIANA2, AIICHIRO KOMORI1, KAZUO ISOBE1, SHIN DEGUCHI1†, MASAYA NISHIYAMA1‡, HIDEYUKI SHIMIZU3, and KEISHI SENOO1 1Department of Applied Biological Chemistry, Graduate School of Agricultural and Life Sciences, The University of Tokyo, 1–1–1 Yayoi, Bunkyo-ku, Tokyo 113–8657, Japan; 2Research Centre for Biology, The Indonesian Institute of Sciences (LIPI), Cibinong Science Centre, Jalan Raya Jakarta-Bogor Km. 46, Cibinong 16911, Indonesia; and 3Asian Environment Research Group, National Institute for Environmental Studies, 16–2 Onogawa, Tsukuba, Ibaraki 305–8506, Japan (Received July 18, 2007—Accepted November 22, 2007) The bacterial community structure in soil of a tropical rainforest in East Kalimantan, Indonesia, where forest fires occurred in 1997–1998, was analysed by denaturing gradient gel electrophoresis (DGGE) with soil samples collected from the area in 2001 and 2002. The study sites were composed of a control forest area without fire damage, a lightly- burned forest area, and a heavily-burned forest area. DGGE band patterns showed that there were many common bac- terial taxa across the areas although the vegetation is not the same. In addition, it was indicated that a change of vegeta- tion in burned areas brought the change in bacterial community structure during 2001–2002. It was also indicated that, depending on a perspective, community structure of soil bacteria in post-fire non-climax forest several years after fire can be more heterogeneous compared with that in unburned climax forest. -
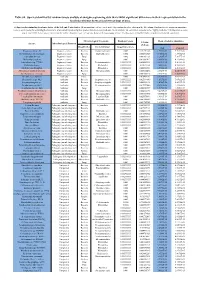
Table S8. Species Identified by Random Forests Analysis of Shotgun Sequencing Data That Exhibit Significant Differences In
Table S8. Species identified by random forests analysis of shotgun sequencing data that exhibit significant differences in their representation in the fecal microbiomes between each two groups of mice. (a) Species discriminating fecal microbiota of the Soil and Control mice. Mean importance of species identified by random forest are shown in the 5th column. Random forests assigns an importance score to each species by estimating the increase in error caused by removing that species from the set of predictors. In our analysis, we considered a species to be “highly predictive” if its importance score was at least 0.001. T-test was performed for the relative abundances of each species between the two groups of mice. P-values were at least 0.05 to be considered statistically significant. Microbiological Taxonomy Random Forests Mean of relative abundance P-Value Species Microbiological Function (T-Test) Classification Bacterial Order Importance Score Soil Control Rhodococcus sp. 2G Engineered strain Bacteria Corynebacteriales 0.002 5.73791E-05 1.9325E-05 9.3737E-06 Herminiimonas arsenitoxidans Engineered strain Bacteria Burkholderiales 0.002 0.005112829 7.1580E-05 1.3995E-05 Aspergillus ibericus Engineered strain Fungi 0.002 0.001061181 9.2368E-05 7.3057E-05 Dichomitus squalens Engineered strain Fungi 0.002 0.018887472 8.0887E-05 4.1254E-05 Acinetobacter sp. TTH0-4 Engineered strain Bacteria Pseudomonadales 0.001333333 0.025523638 2.2311E-05 8.2612E-06 Rhizobium tropici Engineered strain Bacteria Rhizobiales 0.001333333 0.02079554 7.0081E-05 4.2000E-05 Methylocystis bryophila Engineered strain Bacteria Rhizobiales 0.001333333 0.006513543 3.5401E-05 2.2044E-05 Alteromonas naphthalenivorans Engineered strain Bacteria Alteromonadales 0.001 0.000660472 2.0747E-05 4.6463E-05 Saccharomyces cerevisiae Engineered strain Fungi 0.001 0.002980726 3.9901E-05 7.3043E-05 Bacillus phage Belinda Antibiotic Phage 0.002 0.016409765 6.8789E-07 6.0681E-08 Streptomyces sp. -

A002 Methylobacterium Carri Sp. Nov., Isolated from Automotive Air
A002 Methylobacterium carri sp. nov., Isolated from Automotive Air Conditioning System Jigwan Son and Jong-Ok Ka* Department of Agricultural Biotechnology and Research Institute of Agriculture and Life Sciences, Seoul National University A bacterial strain, designated DB0501T, with Gram-stain-negative, aerobic, motile, and rod-shaped cell, was isolated from an automotive air conditioning system collected in the Republic of Korea. 16S rRNA gene sequence analysis indicated that the strain DB0501T grouped in the genus Methylobacterium and closely related to Methylobacterium platani PMB02T (98.8%), Methylobacterium currus PR1016AT (97.7%), Methylobacterium variabile DSM 16961T (97.7%), Methylobacterium aquaticum DSM 16371T (97.6%), Methylobacterium tarhaniae N4211T (97.4%) and Methylobacterium frigidaeris IER25-16T (97.2%). Genomic relatedness between strain DB0501T and its closest relatives was evaluated using average nucleotide identity, digital DNA-DNA hybridization and average amino acid identity with values of 86.4–90.8%, 39.3 ± 2.6–48.2 ± 5.0% and 87.8–89.5% respectively. The strain grew 15-30°C , pH 5.5-8.0 and in 0–1.0% w/v NaCl. Summed feature 3 (C16:1 7c and/or C16:1 6c) and summed feature 8 (C18:1 ω7c T and/or C18:1 ω6c) were the predominant cellular fatty acids in strain DB0501 . Q-10 was the major ubiquinone. The major polar lipids were phosphatidylethanolamine, phosphatidylglycerol, and phosphatidylcholine. The DNA G+C content of strain DB0501T was 70.8 mol%. Based on phenotypic, genotypic and chemotaxonomic data, strain DB0501T represents a novel species of the genus Methylobacterium, for which the name Methylobacterium carri sp. -

Ramlibacter Alkalitolerans Sp. Nov., Alkali-Tolerant Bacterium Isolated from Soil of Ginseng
TAXONOMIC DESCRIPTION Lee and Cha, Int J Syst Evol Microbiol 2017;67:4619–4623 DOI 10.1099/ijsem.0.002342 Ramlibacter alkalitolerans sp. nov., alkali-tolerant bacterium isolated from soil of ginseng Do-Hoon Lee and Chang-Jun Cha* Abstract A novel bacterial strain, designated CJ661T, was isolated from soil of ginseng in Anseong, South Korea. Cells of strain CJ661T were white-coloured, Gram-staining-negative, non-motile, aerobic and rod-shaped. Strain CJ661T grew optimally at 30 C and pH 7.0. The analysis of 16S rRNA gene sequence of strain CJ661T showed that it belongs to the genus Ramlibacter within the family Comamonadaceae and was most closely related to Ramlibacter ginsenosidimutans KCTC 22276T (98.1 %), followed by Ramlibacter henchirensis DSM 14656T (97.1 %). DNA–DNA relatedness levels of strain CJ661T were 40.6 % to R. ginsenosidimutans KCTC 22276T and 25.0 % to R. henchirensis DSM 14656T. The major isoprenoid quinone was ubiquinone (Q-8). The predominant polar lipids were phosphatidylethanolamine, diphosphatidylglycerol and phosphatidylglycerol. The T major cellular fatty acids of strain CJ661 were summed feature 3 (C16 : 1 !6c and/or C16 : 1 !7c), C16 : 0 and summed feature 8 (C18 : 1 !7c and/or C18 : 1 !6c). The G+C content of the genomic DNA was 65.4 mol%. On the basis polyphasic taxonomic data, strain CJ661T represents a novel species in the genus Ramlibacter, for which name Ramlibacter alkalitolerans sp. nov. is proposed; the type strain is CJ661T (=KACC 19305T=JCM 32081T). The genus Ramlibacter was introduced by Heulin et al. [1], (Qiagen). The 16S rRNA gene sequence was determined at and belongs to the family Comamonadaceae in the class Solgent (Daejeon, Korea) using the BigDye Terminator Cycle Betaproteobacteria.