Prediction and Characterization of Therapeutic Protein Aggregation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Caietele CNSAS, Nr. 2 (20) / 2017
Caietele CNSAS Revistă semestrială editată de Consiliul Naţional pentru Studierea Arhivelor Securităţii Minoritatea evreiască din România (I) Anul X, nr. 2 (20)/2017 Editura CNSAS Bucureşti 2018 Consiliul Naţional pentru Studierea Arhivelor Securităţii Bucureşti, str. Matei Basarab, nr. 55-57, sector 3 www.cnsas.ro Caietele CNSAS, anul X, nr. 2 (20)/2017 ISSN: 1844-6590 Consiliu ştiinţific: Dennis Deletant (University College London) Łukasz Kamiński (University of Wroclaw) Gail Kligman (University of California, Los Angeles) Dragoş Petrescu (University of Bucharest & CNSAS) Vladimir Tismăneanu (University of Maryland, College Park) Virgiliu-Leon Ţârău (Babeş-Bolyai University & CNSAS) Katherine Verdery (The City University of New York) Pavel Žáček (Institute for the Study of Totalitarian Regimes, Prague) Colegiul de redacţie: Elis Pleșa (coordonator număr tematic) Liviu Bejenaru Silviu B. Moldovan Liviu Ţăranu (editor) Coperta: Cătălin Mândrilă Machetare computerizată: Liviu Ţăranu Rezumate și corectură text în limba engleză: Gabriela Toma Responsabilitatea pentru conţinutul materialelor aparţine autorilor. Editura Consiliului Naţional pentru Studierea Arhivelor Securităţii e-mail: [email protected] CUPRINS I. Studii Natalia LAZĂR, Evreitate, antisemitism și aliya. Interviu cu Liviu Rotman, prof. univ. S.N.S.P.A. (1 decembrie 2017)………………………………7 Lya BENJAMIN, Ordinul B’nei Brith în România (I.O.B.B.). O scurtă istorie …………………………………………………………………............................25 Florin C. STAN, Aspecte privind emigrarea evreilor din U.R.S.S. -

CITY MANAGER CITY of CAPE Co~
CITY MANAGER CITY OF CAPE co~. DEPARTMENT OF COMMUNITY D~~~aPMi=tfff 3: ftO MEMORANDUM TO: John Szerlag, City Manager FROM: Vincent A. Cautero, Community Develop~-n~.t Director{!t';)\__, Robert H. Pederson, Planning Manager~ Wyatt Daltry, Planning Team Coordinator vl> DATE: September 6, 2016 SUBJECT: Future Land Use Map Amendment Request-LU16-0012 The City has initiated a large scale future land use map amendment for a large area in Northern Cape Coral; the proposed area is 2,818.49 acres. This request is a follow-up to LU15-0004, which brought over 4,000-acres from the Urban Services Reserve Area into the Urban Services Transition Area. Once the amendment is adopted by Council, property owners could rezone their property for development to permit densities supported by centralized water and sewer utilities. The proposed amendment request includes the following: Current FLU Proposed FLU Acreage Single Family/Multi-Family by PDP (SM) SinQle-Family Residential (SF) 2,686.04 SM Multi-Family Residential (MF) 63.16 SM Parks and Recreation (PK) 10.24 Commercial Activity Center (CAC) SF 29.39 CAC MF 29.66 Thank you for your consideration of this future land use map amendment. Please contact Wyatt Daltry, Planning Team Coordinator, at 573-3160 if you have any questions. VAC/wad(North1 +2FLUMAmemoofintent) Attachment Planning Division Case Report LU 16-0012 Review Date: November 2, 2016 Applicant: City of Cape Coral, Department of Community Development Property Owners: See Attachment A Site Address: See Attachment A Authorized Representative: Wyatt Daltry, AICP Planning Team Coordinator City of Cape Coral Department of Community Development (239) 573-3160 Case Staff: Wyatt Daltry, AICP, Planning Team Coordinator Review Approved By: Robert Pederson, AICP, Planning Manager Purpose: The City has initiated this large-scale future land use map amendment for a large area in Northern Cape Coral. -
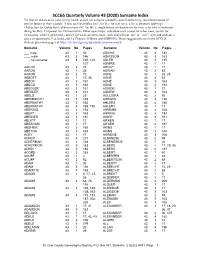
Surname Index to Find All Instances of Your Family Name, Search for Variants Caused by Poor Handwriting, Misinterpretation of Similar Letters Or Their Sounds
SCCGS Quarterly Volume 43 (2020) Surname Index To find all instances of your family name, search for variants caused by poor handwriting, misinterpretation of similar letters or their sounds. A few such examples are L for S, c for e, n for u, u for a; phonetic spellings (Aubuchon for Oubuchon); abbreviations (M’ for Mc ); single letters for double (m for mm, n for nn); translations (King for Roy, Carpenter for Zimmermann). Other search tips: substitute each vowel for other ones, search for nicknames, when hyphenated – search for each surname alone, with and without “de” or “von”; with and without a space or apostrophe (Lachance and La Chance, O’Brien and OBRIEN). More suggestions are on the SCCGS website Quarterly pages at https://stclair-ilgs.org/quarterly-surname-index/ Surname Volume No Pages Surname Volume No Pages ___ male 43 1 50 ADKINS 43 4 182 ___, male 43 3 146 ADKISSON 43 3 163 ___, no surname 43 3 120, 123, ADLER 43 3 135 127 ADMIRE 43 1 17 AACOX 43 2 74 ADOLF 43 1 17 AARON 43 1 25 ADRIAN 43 2 83 AARON 43 2 72 AGNE 43 1 24, 25 ABBOTT 43 1 17, 36 AGNE 43 2 62 ABECK 43 3 161 AGNE 43 3 163 ABEGG 43 3 163 AGNE 43 4 184 ABEGLER 43 3 161 AGNEW 43 1 31 ABEKLER 43 3 161 AGNEW 43 3 163 ABELS 43 1 25 AGULIERA 43 2 93 ABENDROTH 43 3 163 AHEARN 43 3 136 ABERNATHY 43 3 163 AHLERS 43 4 180 ABERNATHY 43 4 185, 190 AHLERT 43 1 17 ABERSOL 43 3 158 AHMANN 43 2 108 ABERT 43 3 163 AHRING 43 4 184 ABIGNER 43 3 161 AIGRE 43 3 161 ABLETT 43 1 17 AITKEN 43 1 17 ABRAHAM 43 1 17 AITKEN 43 4 197 ABSHIER 43 3 169 AKINS 43 1 17 ABSTON 43 3 163 AKINS 43 -

Index to the Case Files of the AJDC Emigration Service, Prague Office, 1945-1950
Index to the Case Files of the AJDC Emigration Service, Prague Office, 1945-1950 This index provides the names of clients served by the AJDC Emigration Service in Czechoslovakia in the years immediately following the end of World War II. It represents the contents of boxes 1-191 of the AJDC Prague Office Collection, held at the Institute for the Study of Totalitarian Regimes, Prague. The JDC Archives received a set of digital files of this collection in 2019 via the U.S. Holocaust Memorial Museum with the Institute’s agreement. The index was created thanks to a group of JDC Archives Indexing Project volunteers and staff. Users of this index are encouraged to try alternate spellings for names (e.g., Ackerman/Ackermann; Lowy/Loewy; Schwartz/Schwarz/Swarc/Swarz). Note that women’s surnames may or may not include the suffix -ova. The “find” feature (PC: ctrl+F; Mac: command+F) may be used to search for names listed in the Additional Name(s) column that may be separated from their alphabetical order. As case files contain private personal information, they are not open to the public. However, family members may request copies of the documents via our online Request Information form. Client Name Additional Name(s) Folder Client Name Additional Name(s) Folder Abeles, Bedrich Marie Abeles, Jiri Abeles 191_04 Abrahamova, 4_56 Abeles, Bedrich 191_10 Veronika Abrahamovic, David Ruzena Abrahamovic, Arnost 5_02 Abeles, Ernest Regina Abeles 4_13 Abrahamovic Abeles, Karel 1_02 Abeles, Kurt 1_03 Abrahamovic, Evzen 1_17 Abeles, Kurt Alice Abeles, Edith -

Monitorul Oficial Dina Treiazi
Anul CXIII Nr. 39. 50 LEI EXEMPLARUL' Sânlbátä, 17 Februarie 1945. REG ATUL ROMANIEI MONITORULPARTEA I a OFICIAL LEGT, DECRETE )URMALE ALE CONSILILILUI DE MINISTRI, DECIZIUNI MINISTERIALE, COMUNICATE, ANUNTURI JUDICIARE (DE INTERES GENERAL) Deshaterlle Partea 1-1 sau a II-a parlement. I LIMA A 3 O N A M E N T E . N L E I P U B L Ie: A T II I NL E T latertla 1 an 16luni3 Zuni I 1 luttasesi une II. O. 'IN- TA.';A: JURNALELE CONSILIULUI DE M1N15TR1: Pa ticu :ari 10.0005.000 2.500 1.200 6.000 Acordarca avantajelor Iegii pentru incurajarca industriel Anioritati de Stat. judet la industriasi si fabricanti 20000 si comune ur- Idem la meseriasi si mori taranesti 4.000 bane 8 000 4.000 2.000 6.000 Schimbari de nome, incetatenirì 8 000 Autoritatl comuaalc rurale 4 000 2.000 1.000 6.000 Comunicate pentru depunerca juramantului de incetatenire 8.000 Autorizari pentru infiintari de birouri vamale 5.000 IN ST.RAINATATE 25.000 12.000 CITATIILF una luna 2500 Curtilor dc Casalte, de Conturi, de ApelstTribunalelor. 1.500 Judecatorillor 750 Un exemplar din anul curent lei 50 part. 1-a60 part. II -a 60 PUBLiCATIILE PENTRU AMENAJAMENTE DE PADURI a . anii trecuti 100 100 Pitduri "titre C 10 ha . tA0 . 10,01 20. 1500 Abonamentul la partcaIII -a (Desbaterile parlamentare) pentru sesittttile. 20,01 100 , 2,500 ordinare. prelungite sau exlraordinare se rocoteste 1.200 lei lunar (minimum 100,01 200 10 009 I luna). w 200,01 500 15.000 Abonamentele rublicaliilerentra autoritari$1 particulars se platesc . -
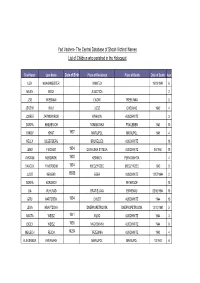
The Central Database of Shoah Victims' Names List of Children Who
Yad Vashem- The Central Database of Shoah Victims' Names List of Children who perished in the Holocaust First Name Last Name Date of Birth Place of Residence Place of Death Date of Death Age LIZA KHAKHMEISTER VINNITZA 19/09/1941 6 MILEN BECK SUBOTICA 2 JZIO WIESMAN LWOW TREBLINKA 8 JERZYK WILK LODZ CHELMNO 1942 4 JOSELE ZARNOWIECKI KRAKOW AUSCHWITZ 3 SONYA KHILKEVICH ROMANOVKA FRAILEBEN 1941 10 YAKOV KHAIT 1937 MARIUPOL MARIUPOL 1941 4 NELLY SILBERBERG BRUXELLES AUSCHWITZ 10 JENO FISCHER 1934 DUNAJSKA STREDA AUSCHWITZ 06/1944 10 AVRAAM NERDINSKI 1938 KISHINEV PERVOMAYSK 4 YAACOV YAVERBOIM 1934 MIEDZYRZEC MIEDZYRZEC 1942 9 JUDIT KEMENY 15555 EGER AUSCHWITZ 12/07/1944 2 SONYA KOROBOV PETERGOF 10 LIA MUHLRAD BRATISLAVA BIRKENAU 05/10/1944 10 GITU HARTSTEIN 1934 CHUST AUSCHWITZ 1944 10 LENA KRAVTZOVA DNEPROPETROVSK DNEPROPETROVSK 13/10/1941 5 AGOTA WEISZ 1941 IKLOD AUSCHWITZ 1944 3 IBOLY WEISZ 1936 NAGYBANYA AUSCHWITZ 1944 8 MEILECH REICH 14254 TRZEBINIA AUSCHWITZ 1943 4 ALEKSANDR AVERBAKH MARIUPOL MARIUPOL 10/1941 6 Yad Vashem- The Central Database of Shoah Victims' Names List of Children who perished in the Holocaust First Name Last Name Date of Birth Place of Residence Place of Death Date of Death Age SALOMON LOTERSPIL 1941 LUBLIN LUBLIN 2 MISHA GORLACHOV 1932 RYASNA RYASNA 1941 9 YEVGENI PUPKIN NOVY OSKOL NOVY OSKOL 4 REVEKKA KRUK 1934 ODESSA ODESSA 1941 7 VOVA ZAYONCHIK 1936 KIEV BABI YAR 29/09/1941 5 MAYA LABOVSKAYA 1931 KIYEV BABI YAR 1941 10 MILOS PLATOVSKY 1932 PROBULOW 1942 10 VILIAM HOENIG 15/12/1939 TREBISOV SACHSENHAUSEN 12/11/1944 -
MMS 260 Spread.P65
Turkish Portuguese Important notes for users in the U.K. Multimedia Speak er System 1. Beyazla iþaretlenmiþ kabloyu beyaz Ön çýkýþ (Front out) 1. Ligue o cabo branco ao conector da tomada “Front out” de 3,5 Mains plug 3,5 jak konektörüne takýn branco This apparatus is fitted with an approved 13 Amp plug. 2. Turuncuyla iþaretlenmiþ kabloyu Turuncu Surround çýkýþ 2. Ligue o cabo laranja ao conector “Surround out” de 3,5 laranja To change a fuse in this type of plug proceed as follows: (Surround out) 3,5 konektörüne takýn 1 Remove fuse cover and fuse. MMS 260 3. Ligue o conector mini Din preto à entrada Din “Control in” MMS 260 3. Siyah mini Din konektörünü Kontrol giriþ (Control in) Din 2 Fix new fuse which should be a BS1362 5 Amp, A.S.T.A. or BSI approved type. 4. Ligue o(s) cabo(s) Line in à fonte de som 3 Refit the fuse cover. giriþine takýn 5. Ligue a fonte de alimentação à entrada “Power in” 4. Hat giriþ (Line in) kablolarýný ses kaynaðýna takýn 6. Ligue o cabo de alimentação à corrente eléctrica If the fitted plug is not suitable for your socket outlets, it should be cut off and an appropriate plug fitted in its place. If the mains plug contains a fuse, this should 5. Güç kaynaðýný Güç giriþ (Power in) giriþine takýn. 7. Ligue/desligue o sistema utilizando o botão “Power/volume” no have a value of 5 Amp. If a plug without a fuse is used, the fuse at the distribution board should not be greater than 5 Amp. -

Alphabetical List of Family Files KEY: * = Book
Alphabetical list of family files Grems-Doolittle Library, Schenectady County Historical Society, 32 Washington Avenue, Schenectady, NY 12305 AAKUS ACKLER ALDA AALTO ACKNER ALDI ABAK ACOME ALDEN ABAR ADAIR ALDRICH ABARE ADAMEC ALDRIDGE ABBA ADAMS* ALESKIEWIZZ ABBALE ADAMSON ALESSANDRINI ABBATIELLO ADLE ALEXANDER ABBATO ADRIANCE ALEXANDERSON p ABBE ADSIT ALEXANDROWICZ ABBEY AERNECKE ALGER*d ABBITT AFFINITO ALHEIM ABBOTT AGAN-AGANS ALINGER ABBS AGARD ALKEMA ABBUHL AHEARN-AHERN ALLAN d ABDALLA AHREET ALLEN p ABEAR AIKEN ALLER ABEEL-ABELL* AINSLIE ALLING ABEL-ABELE AINSWORTH ALLIS ABERBACH AIRD ALMY ABERCROMBIE AKEN-AKIN ALPER ABRAHAM- AKULA ALPHER ABRAHAMS ALBER ALSDORF ALBERS ALSTON ABRAM-ABRAMS ALBERT ALTER ABREU ALBERTI ALTERI-ALTIERI ACHILLES ALBERTS ALTROCK ACKER-AKKER VANDEN AKKER ALBOHM ALVORD ACKERMAN-d* ALBRIGHT-VON (d) AMENT-AMEN- AKKERMAN ALBRICHSFELDT AMANS-AMONS ACKERKNECHT ALBRO AMBLER KEY: * = book; + = book only, no family file; cd = see CD collection; d = see family documents; m = manuscript/personal papers; p = photographs. Alphabetical list of family files Grems-Doolittle Library, Schenectady County Historical Society, 32 Washington Avenue, Schenectady, NY 12305 AMES* APPLETON ASSELSTYNE(see ESSELSTYNE- AMEDORE APPS p ESSELSTINE) AMELL ARAGONA ASTON-ASHTON AMESDEN ARAGOSA AMMERMAN- ARCHER ATHERTON AMERMAN ARCHIBALD ATTANASIO AMON-AMMON ARDELL ATTENDORN AMOS+* ARGERSINGER ATUESTA AMYOT ARGINTEANU ATWELL ANDERS ARKELL ATWOOD ANDERSON p ARKENBURGH AUBE ANDING ARMBRUST AUBREY ANDRES ARMER AUCHAMPAUGH ANDREWS p- ARMITAGE AUDET*-AUDETTE -

Macrourus Berglax Antimora Rostrata Argentina Silus
DEEP-SEA SPECIES BG Дълбоководни видове ES Especies de aguas profundas CS Hlubokomořské druhy DA Dybhavsarter DE Tiefseearten ET Süvaveeliigid EL Είδη βαθέων υδάτων FR Espèces d’eaux profondes GA Speicis Domhainfharraige IT Specie di acque profonde LV Dziļūdens sugas LT Giliavandenės žuvys HU Mélytengeri fajok MT Ħut tal-ibħra fondi NL Diepzeesoorten PL Gatunki głębokowodne PT Espécies de profundidade RO Specii de adâncime SK Hlbokomorské druhy SL Globokomorske vrste FI Syvänmeren lajit SV Djuphavsfisk Lepidopus caudatus Conger conger Polyprion americanus Dalatias licha Molva molva Galeorhinus galeus Centrophorus squamosus Molva dypterygia Centroscymnus coelolepsis Reinhardtius hippoglossoides Phycis spp. Chimaera monstrosa Deania calceus Brosme brosme Coryphaenoides rupestris Centroscyllium fabricii Aphanopus carbo Macrourus berglax Etmopterus spinax Mora moro Galeus melastomus Pagellus bogaraveo Helicolenus dactylopterus Hoplostethus atlanticus Chaceon (Geryon) quinquedens Beryx spp. Apristuris spp. Antimora rostrata Argentina silus Sebastes mentella Pandalus borealis KL-31-13-791-D2-P KL-31-13-791-D2-P Antimora rostrata Aphanopus carbo Apristuris spp. Argentina silus Beryx spp. Brosme brosme Centrophorus Centroscyllium Centroscymnus Chaceon (Geryon) Chimaera Conger conger Coryphaenoides Dalatias licha Deania calceus Etmopterus spinax squamosus fabricii coelolepsis quinquedens monstrosa rupestris BG Исландска котешка Голяма сребърна Португалска Дълбоководен Синя антимора Черна риба сабя акула корюшка Берикс Менек Сива късошипа акула