The Most Developmentally Truncated Fishes Show Extensive Hox Gene
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Puntius Snyderi ERSS
Puntius snyderi (a fish, no common name) Ecological Risk Screening Summary U.S. Fish & Wildlife Service, February 2013 Revised, February 2019 Web Version, 8/8/2019 1 Native Range and Status in the United States Native Range From Chang et al. (2006): “Puntius snyderi is a freshwater cyprinid fish discovered by Oshima when he collected the freshwater fishes in Taiwan in 1915-1917. It was mainly distributed in northern and central Taiwan [Oshima 1919] […].” From Chang et al. (2009): “A similar inference was also proposed for Puntius snyderi and P. semifasciolatus in which P. snyderi is a species endemic to Taiwan and P. semifasciolatus is distributed in both China and Taiwan (Chang et al. 2006). These 2 species were proposed to have differentiated in China. After P. snyderi and P. semifasciolatus dispersed to Taiwan, P. snyderi became extinct in China (Chang et al. 2006).” 1 Chen et al. (2013) list Puntius snyderi as previously present on Kinmen Island, Taiwan but that it is currently locally extinct there. Status in the United States No records of Puntius snyderi in the wild or in trade in the United States were found. Means of Introductions in the United States No records of Puntius snyderi in the wild in the United States were found. Remarks No additional remarks. 2 Biology and Ecology Taxonomic Hierarchy and Taxonomic Standing According to Fricke et al. (2019), Puntius snyderi (Oshima 1919) is the current valid and original name of this species. From Bailly (2017): “Biota > Animalia (Kingdom) > Chordata (Phylum) > Vertebrata (Subphylum) > Gnathostomata (Superclass) > […] Actinopterygii (Class) > Cypriniformes (Order) > Cyprinidae (Family) > Barbinae (Subfamily) > Puntius (Genus) > Puntius snyderi (Species)” Some sources refer to this species by a synonym, Barboides snyderi (Forese and Pauly 2019). -

Molecular Investigations of the Diversity of Freshwater Fishes Across Three Continents
Molecular Investigations of the Diversity of Freshwater Fishes across Three Continents by Malorie M. Hayes A dissertation submitted to the Graduate Faculty of Auburn University in partial fulfillment of the requirements for the Degree of Doctor of Philosophy Auburn, Alabama August 8, 2020 Keywords: Enteromius, Barbus, sub-Saharan Africa, phylogenetics, systematics, Pteronotropis, conservation genetics, Trichomycterus, Guyana Copyright 2020 by Malorie M. Hayes Approved by Jonathan W. Armbruster, Chair, Professor and Director Auburn University Museum of Natural History Department of Biological Sciences Jason E. Bond, Professor and Schlinger Chair in Insect Systematics University of California, Davis Scott R. Santos, Professor and Chair of the Department of Biological Sciences at Auburn University John P. Friel, Director of the Alabama Museum of Natural History Abstract Fishes are the most speciose vertebrates, and incredible diversity can be found within different groups of fish. Due to their physiological limitations, fish are confined to waters, and in freshwater fish, this is restricted to lakes, rivers, and streams. With a constrained habitat like a freshwater system, it can be expected that freshwater fish will show varying levels of diversity depending on a suite of characteristics. Within this dissertation, I examine the diversity of three fish groups: the speciose Enteromius of West Africa, the population genetic diversity of Pteronotropis euryzonus in Alabama and Georgia, and the unexpectedly species rich Trichomycterus from the Guyana highlands. I use molecular methods and geometric morphometrics to determine the systematics of the species and uncover the hidden diversity within their respective groups. When it comes to diversity, the small barbs of Africa are vastly understudied and require a taxonomic revision. -

A Guide to the Parasites of African Freshwater Fishes
A Guide to the Parasites of African Freshwater Fishes Edited by T. Scholz, M.P.M. Vanhove, N. Smit, Z. Jayasundera & M. Gelnar Volume 18 (2018) Chapter 2.1. FISH DIVERSITY AND ECOLOGY Martin REICHARD Diversity of fshes in Africa Fishes are the most taxonomically diverse group of vertebrates and Africa shares a large portion of this diversity. This is due to its rich geological history – being a part of Gondwana, it shares taxa with the Neotropical region, whereas recent close geographical affnity to Eurasia permitted faunal exchange with European and Asian taxa. At the same time, relative isolation and the complex climatic and geological history of Africa enabled major diversifcation within the continent. The taxonomic diversity of African freshwater fshes is associated with functional and ecological diversity. While freshwater habitats form a tiny fraction of the total surface of aquatic habitats compared with the marine environment, most teleost fsh diversity occurs in fresh waters. There are over 3,200 freshwater fsh species in Africa and it is likely several hundreds of species remain undescribed (Snoeks et al. 2011). This high diversity and endemism is likely mirrored in diversity and endemism of their parasites. African fsh diversity includes an ancient group of air-breathing lungfshes (Protopterus spp.). Other taxa are capable of breathing air and tolerate poor water quality, including several clariid catfshes (e.g., Clarias spp.; Fig. 2.1.1D) and anabantids (Ctenopoma spp.). Africa is also home to several bichir species (Polypterus spp.; Fig. 2.1.1A), an ancient fsh group endemic to Africa, and bonytongue Heterotis niloticus (Cuvier, 1829) (Osteoglossidae), a basal actinopterygian fsh. -

Resolving Cypriniformes Relationships Using an Anchored Enrichment Approach Carla C
Stout et al. BMC Evolutionary Biology (2016) 16:244 DOI 10.1186/s12862-016-0819-5 RESEARCH ARTICLE Open Access Resolving Cypriniformes relationships using an anchored enrichment approach Carla C. Stout1*†, Milton Tan1†, Alan R. Lemmon2, Emily Moriarty Lemmon3 and Jonathan W. Armbruster1 Abstract Background: Cypriniformes (minnows, carps, loaches, and suckers) is the largest group of freshwater fishes in the world (~4300 described species). Despite much attention, previous attempts to elucidate relationships using molecular and morphological characters have been incongruent. In this study we present the first phylogenomic analysis using anchored hybrid enrichment for 172 taxa to represent the order (plus three out-group taxa), which is the largest dataset for the order to date (219 loci, 315,288 bp, average locus length of 1011 bp). Results: Concatenation analysis establishes a robust tree with 97 % of nodes at 100 % bootstrap support. Species tree analysis was highly congruent with the concatenation analysis with only two major differences: monophyly of Cobitoidei and placement of Danionidae. Conclusions: Most major clades obtained in prior molecular studies were validated as monophyletic, and we provide robust resolution for the relationships among these clades for the first time. These relationships can be used as a framework for addressing a variety of evolutionary questions (e.g. phylogeography, polyploidization, diversification, trait evolution, comparative genomics) for which Cypriniformes is ideally suited. Keywords: Fish, High-throughput -

BMC Evolutionary Biology Biomed Central
BMC Evolutionary Biology BioMed Central Research article Open Access Evolution of miniaturization and the phylogenetic position of Paedocypris, comprising the world's smallest vertebrate Lukas Rüber*1, Maurice Kottelat2, Heok Hui Tan3, Peter KL Ng3 and Ralf Britz1 Address: 1Department of Zoology, The Natural History Museum, Cromwell Road, London SW7 5BD, UK, 2Route de la Baroche 12, Case postale 57, CH-2952 Cornol, Switzerland (permanent address) and Raffles Museum of Biodiversity Research, National University of Singapore, Kent Ridge, Singapore 119260 and 3Department of Biological Sciences, National University of Singapore, Kent Ridge, Singapore 119260 Email: Lukas Rüber* - [email protected]; Maurice Kottelat - [email protected]; Heok Hui Tan - [email protected]; Peter KL Ng - [email protected]; Ralf Britz - [email protected] * Corresponding author Published: 13 March 2007 Received: 23 October 2006 Accepted: 13 March 2007 BMC Evolutionary Biology 2007, 7:38 doi:10.1186/1471-2148-7-38 This article is available from: http://www.biomedcentral.com/1471-2148/7/38 © 2007 Rüber et al; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Abstract Background: Paedocypris, a highly developmentally truncated fish from peat swamp forests in Southeast Asia, comprises the world's smallest vertebrate. Although clearly a cyprinid fish, a hypothesis about its phylogenetic position among the subfamilies of this largest teleost family, with over 2400 species, does not exist. -

Making a Big Splash with Louisiana Fishes
Making a Big Splash with Louisiana Fishes Written and Designed by Prosanta Chakrabarty, Ph.D., Sophie Warny, Ph.D., and Valerie Derouen LSU Museum of Natural Science To those young people still discovering their love of nature... Note to parents, teachers, instructors, activity coordinators and to all the fishermen in us: This book is a companion piece to Making a Big Splash with Louisiana Fishes, an exhibit at Louisiana State Universi- ty’s Museum of Natural Science (MNS). Located in Foster Hall on the main campus of LSU, this exhibit created in 2012 contains many of the elements discussed in this book. The MNS exhibit hall is open weekdays, from 8 am to 4 pm, when the LSU campus is open. The MNS visits are free of charge, but call our main office at 225-578-2855 to schedule a visit if your group includes 10 or more students. Of course the book can also be enjoyed on its own and we hope that you will enjoy it on your own or with your children or students. The book and exhibit was funded by the Louisiana Board Of Regents, Traditional Enhancement Grant - Education: Mak- ing a Big Splash with Louisiana Fishes: A Three-tiered Education Program and Museum Exhibit. Funding was obtained by LSUMNS Curators’ Sophie Warny and Prosanta Chakrabarty who designed the exhibit with Southwest Museum Services who built it in 2012. The oarfish in the exhibit was created by Carolyn Thome of the Smithsonian, and images exhibited here are from Curator Chakrabarty unless noted elsewhere (see Appendix II). -

(Cypriniformes, Danionidae) in Sundaland Using DNA Barcodes
www.nature.com/scientificreports OPEN Disentangling the taxonomy of the subfamily Rasborinae (Cypriniformes, Danionidae) in Sundaland using DNA barcodes Arni Sholihah1,2, Erwan Delrieu-Trottin2,3, Tedjo Sukmono4, Hadi Dahruddin2,5, Renny Risdawati6, Roza Elvyra7, Arif Wibowo8,9, Kustiati Kustiati10, Frédéric Busson2,11, Sopian Sauri5, Ujang Nurhaman5, Edmond Dounias12, Muhamad Syamsul Arifn Zein5, Yuli Fitriana5, Ilham Vemendra Utama5, Zainal Abidin Muchlisin13, Jean-François Agnèse2, Robert Hanner14, Daisy Wowor5, Dirk Steinke14, Philippe Keith11, Lukas Rüber15,16 & Nicolas Hubert2* Sundaland constitutes one of the largest and most threatened biodiversity hotspots; however, our understanding of its biodiversity is aficted by knowledge gaps in taxonomy and distribution patterns. The subfamily Rasborinae is the most diversifed group of freshwater fshes in Sundaland. Uncertainties in their taxonomy and systematics have constrained its use as a model in evolutionary studies. Here, we established a DNA barcode reference library of the Rasborinae in Sundaland to examine species boundaries and range distributions through DNA-based species delimitation methods. A checklist of the Rasborinae of Sundaland was compiled based on online catalogs and used to estimate the taxonomic coverage of the present study. We generated a total of 991 DNA barcodes from 189 sampling sites in Sundaland. Together with 106 previously published sequences, we subsequently assembled a reference library of 1097 sequences that covers 65 taxa, including 61 of the 79 known Rasborinae species of Sundaland. Our library indicates that Rasborinae species are defned by distinct molecular lineages that are captured by species delimitation methods. A large overlap between intraspecifc and interspecifc genetic distance is observed that can be explained by the large amounts of cryptic diversity as evidenced by the 166 Operational Taxonomic Units detected. -

'Barbus' (Cyprinidae)
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Texas A&M University GENETIC CHARACTERIZATION OF THE GENUS ‘BARBUS’ (CYPRINIDAE) IN THE LAKE VICTORIA DRAINAGE SYSTEM, KENYA A Thesis by VIOLET M’MBONE NDEDA Submitted to the Office of Graduate and Professional Studies of Texas A&M University in partial fulfillment of the requirements for the degree of MASTER OF SCIENCE Chair of Committee, Luis A. Hurtado Co-Chair of Committee, Mariana Mateos Committee Member, James B. Woolley Head of Department, David Caldwell December 2018 Major Subject: Wildlife and Fisheries Sciences Copyright 2018 Violet M’mbone Ndeda ABSTRACT The genus ‘Barbus’ belongs to a speciose taxonomically complex and heterogeneous assemblage of cyprinid fish. In Lake Victoria drainage basin in Kenya, ten endemic species of ‘Barbus’ are reported, which play a significant role in food security and socio-economic development of the local community. Although these species are identified using morphological characters, confusion may occur when trying to distinguish morphologically similar species. Recent molecular work in the region has suggested presence of introgression within certain ‘Barbus’ species further complicating the taxonomy and species identification in the group. In this study, we obtained cytochrome b and GH-intron 2 gene sequences of nine ‘Barbus’ species sampled in the Lake Victoria drainage basin in Kenya. We conducted Maximum likelihood and Bayesian phylogenetic analyses to establish their evolutionary relationships in relation to other ‘Barbus’. The results showed distinct lineages of ‘Barbus’ species not subjected to introgression/hybridization. Herein, we present new sequences of cytochrome b and GH DNA for small African ‘Barbus’. -

Cypriniformes of Borneo (Actinopterygii, Otophysi): an Extraordinary Fauna for Integrated Studies on Diversity, Systematics, Evolution, Ecology, and Conservation
Zootaxa 3586: 359–376 (2012) ISSN 1175-5326 (print edition) www.mapress.com/zootaxa/ ZOOTAXA Copyright © 2012 · Magnolia Press Article ISSN 1175-5334 (online edition) urn:lsid:zoobank.org:pub:7A06704C-8DE5-4B9F-9F4B-42F7C6C9B32F Cypriniformes of Borneo (Actinopterygii, Otophysi): An Extraordinary Fauna for Integrated Studies on Diversity, Systematics, Evolution, Ecology, and Conservation ZOHRAH H. SULAIMAN1 & R.L MAYDEN2 1Biological Science Programme, Faculty of Science, Universiti Brunei Darussalam, Tungku BE1410, Brunei Darussalam; E-mail:[email protected] 2Department of Biology, 3507 Laclede Ave, Saint Louis University, St Louis, Missouri 63103, USA; E-mail:[email protected] Abstract Borneo Island is governed by the countries of Brunei Darussalam, Malaysia (Sabah and Sarawak) and Indonesia (Kalimantan) and is part of Sundaland. These countries have a high diversity of freshwater fishes, especially described and undescribed species of Cypriniformes; together these species and other flora and fauna represent an extraordinary opportunity for worldwide collaboration to investigate the biodiversity, conservation, management and evolution of Borneo’s wildlife. Much of the fauna and flora of Borneo is under significant threat, warranting an immediate and swift international collaboration to rapidly inventory, describe, and conserve the diversity. The Sunda drainage appears to have been an important evolutionary centre for many fish groups, including cypriniforms (Cyprinidae, Balitoridae and Gyrinocheilidae); however, Northwestern Borneo (Brunei, Sabah and Sarawak) is not connected to Sundaland, and this disjunction likely explains the non-homogeneity of Bornean ichthyofauna. A previous study confirmed that northern Borneo, eastern Borneo and Sarawak shared a similar ichthyofauna, findings that support the general hypothesis for freshwater connections at one time between western Borneo and central Sumatra, and south Borneo and Java island. -
Reply to Yang Et Al. 2015
See discussions, stats, and author profiles for this publication at: http://www.researchgate.net/publication/278047497 Nomenclatural changes should not be based on equivocally supported phylogenies: Reply to Yang et al. 2015 ARTICLE in MOLECULAR PHYLOGENETICS AND EVOLUTION · JUNE 2015 Impact Factor: 3.92 · DOI: 10.1016/j.ympev.2015.05.025 · Source: PubMed READS 88 2 AUTHORS: Ray C. Schmidt Henry Bart Smithsonian Tropical Research Institute Tulane University 7 PUBLICATIONS 1 CITATION 56 PUBLICATIONS 613 CITATIONS SEE PROFILE SEE PROFILE All in-text references underlined in blue are linked to publications on ResearchGate, Available from: Ray C. Schmidt letting you access and read them immediately. Retrieved on: 29 October 2015 Molecular Phylogenetics and Evolution 90 (2015) 193–194 Contents lists available at ScienceDirect Molecular Phylogenetics and Evolution journal homepage: www.elsevier.com/locate/ympev Letter to the Editor Nomenclatural changes should not be based on equivocally supported phylogenies: Reply to Yang et al. 2015 q ⇑ Ray C. Schmidt , Henry L. Bart Jr. Department of Ecology and Evolutionary Biology, Tulane University, 400 Boggs Hall, New Orleans, LA 70118, United States Tulane University Biodiversity Research Institute, Belle Chasse, LA 70037, United States article info abstract Article history: Phylogenies produced by Yang et al. 2015 provide reasonably well-supported hypotheses of relationships Received 22 May 2015 among 11 proposed tribes of cyprinine fishes and present an interesting hypothesis about the origin of a Accepted 31 May 2015 number of polyploid cyprinine lineages. However, support for relationships within some of the tribes is Available online 6 June 2015 equivocal. Herein we address the treatment of African diploid and tetraploid cyprinine genera within tribe Smiliogastrini. -
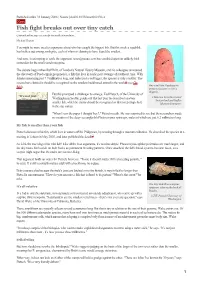
Fish Fight Breaks out Over Tiny Catch
Published online 31 January 2006 | Nature | doi:10.1038/news060130-4 News Fish fight breaks out over tiny catch Contenders line up to net credit for smallest vertebrate. Michael Hopkin You might be more used to arguments about who has caught the biggest fish. But this week a squabble has broken out among zoologists, each of whom is claiming to have found the smallest. And now, in attempting to settle the argument, [email protected] has stumbled upon an unlikely third contender for the small-vertebrate prize. The debate began when Ralf Britz, of London's Natural History Museum, and his colleagues announced the discovery of Paedocypris progenetica, a fish that lives in acidic peat swamps of southeast Asia. With females measuring just 7.9 millimetres long, and males just a tad bigger, the species is truly a tiddler. The researchers claimed it should be recognized as the smallest backboned animal in the world1 (see Go One small fish: Paedocypris fish!). progenetica gets lost on a fingertip. But this prompted a challenger to emerge. Ted Pietsch, of the University of “It's not just Washington in Seattle, points out that last year he described an even © Maurice Kottelat Cornol millimetres that Switzerland and Raffles count - it's how smaller fish, which he claims should be recognized as first (or perhaps last) Museum Singapore you use those in the size stakes. millimetres.” "When I saw the paper I thought 'hey!'," Pietsch recalls. He was surprised to see that the researchers made no mention of the deep-sea anglerfish Photocorynus spiniceps, males of which are just 6.2 millimetres long. -

Zootaxa, Betadevario Ramachandrani, a New Danionine Genus And
Zootaxa 2519: 31–47 (2010) ISSN 1175-5326 (print edition) www.mapress.com/zootaxa/ Article ZOOTAXA Copyright © 2010 · Magnolia Press ISSN 1175-5334 (online edition) Betadevario ramachandrani, a new danionine genus and species from the Western Ghats of India (Teleostei: Cyprinidae: Danioninae) P. K. PRAMOD1, FANG FANG2, K. REMA DEVI3, TE-YU LIAO2,4, T.J. INDRA5, K.S. JAMEELA BEEVI6 & SVEN O. KULLANDER2,7 1Marine Products Export Development Authority, Sri Vinayaka Kripa, Opp. Ananda shetty Circle, Attavar, Mangalore, Karnataka 575 001, India. E-mail: [email protected] 2Department of Vertebrate Zoology, Swedish Museum of Natural History, POB 50007, SE-104 05 Stockholm, Sweden E-mail: [email protected];[email protected]; [email protected] 3Marine Biological Station, Zoological Survey of India, 130, Santhome High Road, Chennai, India 4Department of Zoology, Stockholm University, 106 91 Stockholm, Sweden 5Southern Regional Station, Zoological Survey of India, 130, Santhome High Road, Chennai, India 6Department of Zoology, Maharajas College, Ernukalam, India 7Corresponding author. E-mail: [email protected] Abstract Betadevario, new genus, with the single species B. ramachandrani, new species, from Karnataka, southwestern India, is closely related to Devario but differs from it in having two pairs of long barbels (vs. two pairs of short or rudimentary barbels, or barbels absent), wider cleithral spot which extends to cover three scales horizontally (vs. covering only one scale in width), long and low laminar preorbital process (vs. absent or a slender pointed spine-like process) along the anterior margin of the orbit, a unique flank colour pattern with a wide dark band along the lower side, bordered dorsally by a wide light stripe (vs.