Role of Endog in Development and Cell Injury
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Inhibition of LSD1 Via Sequestration Contributes to Tau-Mediated Neurodegeneration
The inhibition of LSD1 via sequestration contributes to tau-mediated neurodegeneration Amanda K. Engstroma, Alicia C. Walkera, Rohitha A. Moudgala, Dexter A. Myricka, Stephanie M. Kylea, Yu Baia, M. Jordan Rowleyb, and David J. Katza,1 aDepartment of Cell Biology, Emory University School of Medicine, Atlanta, GA 30322; and bDepartment of Genetics, Cell Biology, and Anatomy, University of Nebraska Medical Center, Omaha, NE 68198 Edited by Michele Pagano, HHMI and New York University School of Medicine, New York, NY, and accepted by Editorial Board Member Gail Mandel October 1, 2020 (received for review July 2, 2020) Tauopathies are a class of neurodegenerative diseases associated AD cases, but not other neurodegenerative diseases, such as with pathological tau. Despite many advances in our understand- Parkinson’s disease or amyotrophic lateral sclerosis (ALS) cases. ing of these diseases, the direct mechanism through which tau Consistent with this overlap, we observed LSD1 protein mis- contributes to neurodegeneration remains poorly understood. Pre- localized to cytoplasmic NFTs, but not associated with Aβ plaques viously, our laboratory implicated the histone demethylase LSD1 in in AD cases or Lewy bodies of α-synuclein in Parkinson’sdisease tau-induced neurodegeneration by showing that LSD1 localizes to cases (26). In control cases, LSD1 remains strictly confined to the pathological tau aggregates in Alzheimer’s disease cases, and that it nucleus (26), due to its well-defined nuclear localization signal is continuously required for the survival of hippocampal and cortical (27). These data highlight the requirement for LSD1 in neuro- neurons in mice. Here, we utilize the P301S tauopathy mouse model nal survival and suggest that the nuclear function of the histone to demonstrate that pathological tau can exclude LSD1 from the demethylase LSD1 could be disrupted by mislocalization to nucleus in neurons. -

ENDOG Impacts on Tumor Cell Proliferation and Tumor Prognosis in the Context of PI3K/PTEN Pathway Status
cancers Article ENDOG Impacts on Tumor Cell Proliferation and Tumor Prognosis in the Context of PI3K/PTEN Pathway Status Gisel Barés 1,†, Aida Beà 1,†, Luís Hernández 2,* , Raul Navaridas 3, Isidre Felip 3, Cristina Megino 3 , Natividad Blasco 1,‡, Ferran Nadeu 2 , Elías Campo 2,4, Marta Llovera 1 , Xavier Dolcet 3 and Daniel Sanchis 1,* 1 Departament de Ciències Mèdiques Bàsiques, Universitat de Lleida-IRBLleida, 25198 Lleida, Spain; [email protected] (G.B.); [email protected] (A.B.); [email protected] (N.B.); [email protected] (M.L.) 2 Lymphoid Neoplasm Program, Institut d’Investigacions Biomèdiques Agustí Pi i Sunyer (IDIBAPS) and CIBERONC, 08036 Barcelona, Spain; [email protected] (F.N.); [email protected] (E.C.) 3 Departament de Ciències Mèdiques Bàsiques, Universitat de Lleida–IRBLleida and CIBERONC, 25198 Lleida, Spain; [email protected] (R.N.); [email protected] (I.F.); [email protected] (C.M.); [email protected] (X.D.) 4 Department of Oncology, Hospital Clinic of Barcelona, Universitat de Barcelona, 08036 Barcelona, Spain * Correspondence: [email protected] (L.H.); [email protected] (D.S.); Tel.: +34-(97)-375-2949 (D.S.) † These authors contributed equally to this work (co-first authors). ‡ Present address: Grupo LMA Fundación PETHEMA, Laboratorio de Biología Molecular, Hospital Universitario y Politécnico La Fe, 46026 Valencia, Spain. Simple Summary: The PI3K/AKT pathway is involved in cell survival and proliferation. Molecular aberrations and/or hyperactivation of the PI3K-PTEN-AKT axis are frequent in distinct cancer types such as endometrial carcinoma, the most common type of cancer of the female genital tract, and chronic lymphocytic leukemia (CLL), a mature B-cell neoplasm depending on B-cell receptor (BCR) activity, which induces chronical activation of this pathway. -

Association of Endonuclease G Gene Variants with Cardiovascular Disease Risk Factors
Reports of Biochemistry & Molecular Biology Vol.8, No.2, July 2019 Original article www.RBMB.net Association of Endonuclease G Gene Variants with Cardiovascular Disease Risk Factors Negar Etehad Roodi#1, Nushin Karkuki Osguei#2, Mahdy Hasanzadeh Daloee3, Alireza Pasdar4, Majid Ghayour-Mobarhan5, Gordon Ferns6, Ali Samadi Kuchaksaraei*¹ Abstract Background: Cardiovascular disease (CVD) is a leading cause of death, supporting the need for the identification of novel biomarkers as risk stratification factors. Endonuclease G (ENDOG) has recently been suggested to be a novel determinant of cardiac hypertrophy and mitochondrial function, and plays an important role in apoptosis processes involved in cardiac myocyte death. The aim of current study was to explore the association of two genetic variants in ENDOG gene (ENDOG) with CVD risk factors in an Iranian population. Methods: Subjects included 663 patients with CVD and 282 healthy individuals recruited as part of the Mashhad Stroke and Heart Atherosclerotic Disorders Cohort Study. The ENDOG S12L (rs 2293969) and L142M (rs 61397314) variants were genotyped. Anthropometric and biochemical factors were measured in all the subjects followed by univariate and multivariate analyses to determine the association of these genetic markers with CVD and biochemical parameters. Results: ENDOG polymorphisms were found at a significantly higher prevalence in individuals who had histories of smoking and breaking point in L142M. In contrast, other risk factors for cardiovascular disease, including lipid profile and blood pressure, showed no or very weak relationship with the ENDOG polymorphisms. Conclusions: Our findings indicated an association between an ENDOG genetic variant and smoking history as a cardiovascular risk factor. Further studies in the prospective setting are warranted to investigate the value of this marker. -

Resolvin D3 Is Dysregulated in Arthritis and Reduces Arthritic Inflammation Hildur H
Resolvin D3 Is Dysregulated in Arthritis and Reduces Arthritic Inflammation Hildur H. Arnardottir, Jesmond Dalli, Lucy V. Norling, Romain A. Colas, Mauro Perretti and Charles N. Serhan This information is current as of September 27, 2021. J Immunol published online 17 August 2016 http://www.jimmunol.org/content/early/2016/08/17/jimmun ol.1502268 Downloaded from Supplementary http://www.jimmunol.org/content/suppl/2016/08/17/jimmunol.150226 Material 8.DCSupplemental Why The JI? Submit online. http://www.jimmunol.org/ • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication *average by guest on September 27, 2021 Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2016 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. Published August 17, 2016, doi:10.4049/jimmunol.1502268 The Journal of Immunology Resolvin D3 Is Dysregulated in Arthritis and Reduces Arthritic Inflammation Hildur H. Arnardottir,* Jesmond Dalli,*,1 Lucy V. Norling,† Romain A. Colas,*,1 Mauro Perretti,† and Charles N. Serhan* Uncontrolled inflammation is a unifying component of many chronic inflammatory diseases, such as arthritis. -

Protacs for Treatment of Cancer
0031-3998/10/6705-0505 Vol. 67, No. 5, 2010 PEDIATRIC RESEARCH Printed in U.S.A. Copyright © 2010 International Pediatric Research Foundation, Inc. Protacs for Treatment of Cancer KATHLEEN M. SAKAMOTO Department of Pediatrics, David Geffen School of Medicine at UCLA, Los Angeles, California 90095 ABSTRACT: Protein degradation is the cell’s mechanism of elim- attaches the ubiquitin to a substrate in an ATP-dependent reac- inating misfolded or unwanted proteins. The pathway by which tion. Second, the ubiquitin is transferred to the ubiquitin- proteins are degraded occurs through the ubiquitin-proteasome sys- conjugating enzyme or E2, which transfers the ubiquitin together tem. Ubiquitin is a small 9-kD (kDa) protein that is attached to with an E3 ubiquitin ligase, to the protein (Fig. 1). The E3 proteins. A minimum of four ubiquitins are required for proteins to be ubiquitin ligase confers specificity to the protein target. In the recognized by the degradation machinery, known as the 26S protea- human genome, there is one E1 enzyme, approximately 50 E2 some. Defects in ubiquitination have been identified in a number of diseases, including cancer, neurodegenerative diseases, and meta- enzymes, and more than 600 E3 enzymes (4,5,11,12). bolic disorders. We sought to exploit the delicate balance between Mutations or abnormal regulation of ubiquitin ligases have protein synthesis and degradation to treat cancer by designing a been identified in cancer. In addition, several targets of E3 chimeric molecule, known as Protac (Proteolysis Targeting Chimeric ubiquitin ligases are mutated or are overexpressed in tumors. molecule). Protacs are heterobifunctional nanomolecules that are Among these proteins are the tumor suppressors p53, (breast approximately 10 nm in size and can recruit proteins that cause cancer1, early onset) BRCA1, cyclin inhibitor kinase inhibitor 1B cancer to the ubiquitin-proteasome machinery for degradation. -

Mass Spectrometry-Based Absolute Quantification of 20S Proteasome
Mass Spectrometry-based Absolute Quantification of 20S Proteasome Status for Controlled Ex-vivo Expansion of Human Adipose-derived Mesenchymal Stromal/Stem Cells Thomas Menneteau, Bertrand Fabre, Luc Garrigues, Alexandre Stella, Dusan Zivkovic, Florence Roux-Dalvai, Emmanuelle Mouton-Barbosa, Mathilde Beau, Marie-Laure Renoud, François Amalric, et al. To cite this version: Thomas Menneteau, Bertrand Fabre, Luc Garrigues, Alexandre Stella, Dusan Zivkovic, et al.. Mass Spectrometry-based Absolute Quantification of 20S Proteasome Status for Controlled Ex-vivo Expansion of Human Adipose-derived Mesenchymal Stromal/Stem Cells. Molecular and Cellular Proteomics, American Society for Biochemistry and Molecular Biology, 2019, 18 (4), pp.744-759. 10.1074/mcp.RA118.000958. hal-02167451 HAL Id: hal-02167451 https://hal.archives-ouvertes.fr/hal-02167451 Submitted on 27 Jun 2019 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Research Mass Spectrometry-based Absolute Quantification of 20S Proteasome Status for Controlled Ex-vivo Expansion of Human Adipose-derived -

Role of Specialized Pro-Resolving Lipid Mediators in Pulmonary
Yang et al. Respir Res (2021) 22:204 https://doi.org/10.1186/s12931-021-01792-y REVIEW Open Access Role of specialized pro-resolving lipid mediators in pulmonary infammation diseases: mechanisms and development Ailin Yang, Yanjun Wu, Ganggang Yu* and Haoyan Wang* Abstract Infammation is an essential mechanism of various diseases. The development and resolution of infammation are complex immune-modulation processes which induce the involvement of various types of immune cells. Specialized pro-resolving lipid mediators (SPMs) have been demonstrated to be signaling molecules in infammation. SPMs are involved in the pathophysiology of diferent diseases, especially respiratory diseases, including asthma, pneumonia, and chronic obstructive pulmonary disease. All of these diseases are related to the infammatory response and its persistence. Therefore, a deeper understanding of the mechanisms and development of infammation in respiratory disease, and the roles of the SPM family in the resolution process, might be useful in the quest for novel therapies and preventive measures for pulmonary diseases. Keywords: Infammation resolution, Lung diseases, Lung infammation, Pro-resolving lipid mediators, Mechanism Introduction have shown that chronic, persistent infammation might Infammation is a major defense mechanism of the be the cause of cancer, neurodegenerative diseases [5] human body. If the body is subjected to an injury, infec- (e.g., Alzheimer’s disease, Parkinson disease) and even tion, or other similar stimuli, the innate immune system certain types of mental illness [6]. becomes activated within seconds-to-minutes. Innate Acute infammation is induced by the innate immune immunity is useful in promoting infammation and can system, which has an essential protective role, prevents aid in determining the level of pathogenic invasion or some infections, and promotes healing in injured tis- stimuli. -

Mouse Models of Human Disease. Part I: Techniques and Resources for Genetic Analysis in Mice
Downloaded from genesdev.cshlp.org on September 29, 2021 - Published by Cold Spring Harbor Laboratory Press REVIEW Mouse models of human disease. Part I: Techniques and resources for genetic analysis in mice Mary A. Bedell, 1 Nancy A. Jenkins, and Neal G. Copeland 2 Mammalian Genetics Laboratory, ABL-Basic Research Program, NCI-Frederick Cancer Research and Development Center, Frederick, Maryland 21702-1201 USA. The mouse is an ideal model organism for human dis- >465 (Festing 1994). Inbred strains are produced by at ease. Not only are mice physiologically similar to hu- least 20 consecutive generations of brother-sister mating mans, but a large genetic reservoir of potential models of and were developed originally for the study of cancer human disease has been generated through the identifi- (Morse 1981). However, inbred strains of mice differ in cation of >1000 spontaneous, radiation- or chemically hundreds of other ways besides tumor susceptibility and induced mutant loci. In addition, a number of recent they thus provide models for many other diseases be- technological advances have dramatically increased our sides cancer. ability to create mouse models of human disease. These In the 1940s, George Snell pioneered the development technological advances include the development of high- of a new kind of inbred strain, the congenic strain, to resolution genetic and physical linkage maps of the isolate loci affecting tissue transplantation (for review, mouse genome, which in turn are facilitating the iden- see Flaherty 1981; Frankel 1995). Like the inbred strains, tification and cloning of mouse disease loci. Further- congenic strains have proven extremely useful for dis- more, transgenic technologies that allow one to ectopi- ease research. -

Acne in Adolescence—Protecting the Heart but Damaging the Prostate Later in Life?
American Journal of Epidemiology Vol. 161, No. 12 Copyright ª 2005 by the Johns Hopkins Bloomberg School of Public Health Printed in U.S.A. All rights reserved DOI: 10.1093/aje/kwi148 Invited Commentary: Acne in Adolescence—Protecting the Heart but Damaging the Prostate Later in Life? Earl S. Ford1 and Simin Liu2,3 1 Division of Adult and Community Health, National Center for Chronic Disease Prevention and Health Promotion, Centers for Disease Control and Prevention, Atlanta, GA. 2 Division of Preventive Medicine, Brigham and Women’s Hospital and Harvard Medical School, Boston, MA. 3 Department of Epidemiology, Harvard School of Public Health, Boston, MA. Received for publication February 24, 2005; accepted for publication March 7, 2005. Abbreviation: SHBG, sex hormone-binding globulin. Man is only man at the surface. Remove the skin, dissect, and studies can be creatively used to generate new hypotheses immediately you come to machinery. about the potential pathogenetic pathways of various —Paul Valery, French critic and poet (1871–1945) conditions. Consistent with the longstanding hypothesis that endog- Cutaneous manifestations of diseases have long helped enous sex hormones play important roles in the pathogenesis observant clinicians to establish a diagnosis (1), but can of prostate cancer and cardiovascular disease, the authors’ cutaneous clues or other physical characteristics predict view of acne largely as a manifestation of endogenous hor- the risk of mortality or incident disease far into the future? monal activity, principally androgen activity, has precedent. For some time, researchers have linked with mixed success Although there are many reasons to believe that androgen various physical manifestations to the risk of developing activity may increase the risk of prostate cancer, epidemio- diseases, such as coronary heart disease. -

Trans-Synaptic and Retrograde Axonal Spread of Lewy Pathology Following
Schaser et al. acta neuropathol commun (2020) 8:150 https://doi.org/10.1186/s40478-020-01026-0 RESEARCH Open Access Trans-synaptic and retrograde axonal spread of Lewy pathology following pre-formed fbril injection in an in vivo A53T alpha-synuclein mouse model of synucleinopathy Allison J. Schaser1, Teresa L. Stackhouse1, Leah J. Weston1, Patrick C. Kerstein2, Valerie R. Osterberg1, Claudia S. López3, Dennis W. Dickson4, Kelvin C. Luk5, Charles K. Meshul6,7, Randall L. Woltjer8 and Vivek K. Unni1,9* Abstract It is necessary to develop an understanding of the specifc mechanisms involved in alpha-synuclein aggregation and propagation to develop disease modifying therapies for age-related synucleinopathies, including Parkinson’s disease and Dementia with Lewy Bodies. To adequately address this question, we developed a new transgenic mouse model of synucleinopathy that expresses human A53T SynGFP under control of the mouse prion protein promoter. Our characterization of this mouse line demonstrates that it exhibits several distinct advantages over other, currently avail- able, mouse models. This new model allows rigorous study of the initial location of Lewy pathology formation and propagation in the living brain, and strongly suggests that aggregation begins in axonal structures with retrograde propagation to the cell body. This model also shows expeditious development of alpha-synuclein pathology follow- ing induction with small, in vitro-generated alpha-synuclein pre-formed fbrils (PFFs), as well as accelerated cell death of inclusion-bearing cells. Using this model, we found that aggregated alpha-synuclein somatic inclusions developed frst in neurons, but later showed a second wave of inclusion formation in astrocytes. -
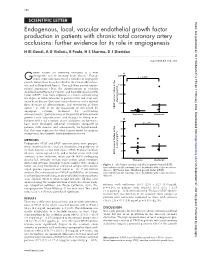
Endogenous, Local, Vascular Endothelial Growth
158 SCIENTIFIC LETTER Heart: first published as 10.1136/heart.87.2.158 on 1 February 2002. Downloaded from Endogenous, local, vascular endothelial growth factor production in patients with chronic total coronary artery occlusions: further evidence for its role in angiogenesis H El-Gendi, A G Violaris, R Foale, H S Sharma, D J Sheridan ............................................................................................................................. Heart 2002;87:158–159 rowth factors are currently emerging as a new A 1 2000 therapeutic tool in coronary heart disease. Endog- p = 0.026 Genous expression patterns of a number of angiogenic growth factors have been described in the chronically ischae- mic and collateralised hearts.2 Through their proven experi- 1500 mental angiogenic effect, the administration of vascular endothelial growth factor (VEGF)3 and basic fibroblast growth factor (bFGF)1 have been explored as a means of increasing 1000 the degree of collateralisation in patients with end stage cor- onary heart disease. Questions remain however on the optimal plasma (pg/ml) 500 dose, duration of administration, and monitoring of these 1–3 agents as well as on the magnitude of risk posed by VEGF concentration in the angiogenic cytokine treatment for accelerating 0 atherosclerosis,45partly because of the paucity of data on local Stenoses Occlusion growth factor concentrations and changes in living man. B Patients with total coronary artery occlusions are known to 50 have more developed collateral circulation compared to p = -

Accelerating the Reversal of Inflammatory Pain with NPD1 and Its Receptor GPR37
Accelerating the reversal of inflammatory pain with NPD1 and its receptor GPR37 Lintao Qu, Michael J. Caterina J Clin Invest. 2018;128(8):3246-3249. https://doi.org/10.1172/JCI122203. Commentary Resolution of inflammation is a critical process that is facilitated by specialized proresolving mediators (SPMs). In this issue, Bang et al. show that the G protein–coupled receptor GPR37 is a receptor for one such SPM, neuroprotectin D1. They also show that GPR37 activation in macrophages enhances phagocytosis, shifts cytokine release toward an antiinflammatory profile, and thereby helps to reverse inflammatory pain. Find the latest version: https://jci.me/122203/pdf COMMENTARY The Journal of Clinical Investigation Accelerating the reversal of inflammatory pain with NPD1 and its receptor GPR37 Lintao Qu1,2 and Michael J. Caterina1,2,3,4 1Department of Neurosurgery, 2Neurosurgery Pain Research Institute, 3Department of Biological Chemistry, and 4Department of Neuroscience, Johns Hopkins School of Medicine, Baltimore, Maryland, USA. NPD1, GPR37, and the resolution of inflammatory pain Resolution of inflammation is a critical process that is facilitated by GPR37 is a GPCR most extensively stud- specialized proresolving mediators (SPMs). In this issue, Bang et al. show ied in the brain that has been associ- that the G protein–coupled receptor GPR37 is a receptor for one such SPM, ated with Parkinson’s disease and autism neuroprotectin D1. They also show that GPR37 activation in macrophages spectrum disorder (14–16). Two known enhances phagocytosis, shifts cytokine release toward an antiinflammatory GPR37 ligands, prosaposin and prosaposin- profile, and thereby helps to reverse inflammatory pain. derived 14-mer peptide (TX14), have been shown to exert neuroprotective and glioprotective effects via this recep- tor (17).