Dissertation.Pdf
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
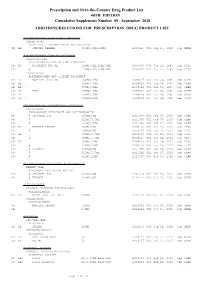
Additions and Deletions to the Drug Product List
Prescription and Over-the-Counter Drug Product List 40TH EDITION Cumulative Supplement Number 09 : September 2020 ADDITIONS/DELETIONS FOR PRESCRIPTION DRUG PRODUCT LIST ACETAMINOPHEN; BUTALBITAL; CAFFEINE TABLET;ORAL BUTALBITAL, ACETAMINOPHEN AND CAFFEINE >A> AA STRIDES PHARMA 325MG;50MG;40MG A 203647 001 Sep 21, 2020 Sep NEWA ACETAMINOPHEN; CODEINE PHOSPHATE SOLUTION;ORAL ACETAMINOPHEN AND CODEINE PHOSPHATE >D> AA WOCKHARDT BIO AG 120MG/5ML;12MG/5ML A 087006 001 Jul 22, 1981 Sep DISC >A> @ 120MG/5ML;12MG/5ML A 087006 001 Jul 22, 1981 Sep DISC TABLET;ORAL ACETAMINOPHEN AND CODEINE PHOSPHATE >A> AA NOSTRUM LABS INC 300MG;15MG A 088627 001 Mar 06, 1985 Sep CAHN >A> AA 300MG;30MG A 088628 001 Mar 06, 1985 Sep CAHN >A> AA ! 300MG;60MG A 088629 001 Mar 06, 1985 Sep CAHN >D> AA TEVA 300MG;15MG A 088627 001 Mar 06, 1985 Sep CAHN >D> AA 300MG;30MG A 088628 001 Mar 06, 1985 Sep CAHN >D> AA ! 300MG;60MG A 088629 001 Mar 06, 1985 Sep CAHN ACETAMINOPHEN; HYDROCODONE BITARTRATE TABLET;ORAL HYDROCODONE BITARTRATE AND ACETAMINOPHEN >A> @ CEROVENE INC 325MG;5MG A 211690 001 Feb 07, 2020 Sep CAHN >A> @ 325MG;7.5MG A 211690 002 Feb 07, 2020 Sep CAHN >A> @ 325MG;10MG A 211690 003 Feb 07, 2020 Sep CAHN >D> AA VINTAGE PHARMS 300MG;5MG A 090415 001 Jan 24, 2011 Sep DISC >A> @ 300MG;5MG A 090415 001 Jan 24, 2011 Sep DISC >D> AA 300MG;7.5MG A 090415 002 Jan 24, 2011 Sep DISC >A> @ 300MG;7.5MG A 090415 002 Jan 24, 2011 Sep DISC >D> AA 300MG;10MG A 090415 003 Jan 24, 2011 Sep DISC >A> @ 300MG;10MG A 090415 003 Jan 24, 2011 Sep DISC >D> @ XIROMED 325MG;5MG A 211690 -

P2 Receptors in Cardiovascular Regulation and Disease
Purinergic Signalling (2008) 4:1–20 DOI 10.1007/s11302-007-9078-7 REVIEW P2 receptors in cardiovascular regulation and disease David Erlinge & Geoffrey Burnstock Received: 3 May 2007 /Accepted: 22 August 2007 /Published online: 21 September 2007 # Springer Science + Business Media B.V. 2007 Abstract The role of ATP as an extracellular signalling Introduction molecule is now well established and evidence is accumulating that ATP and other nucleotides (ADP, UTP and UDP) play Ever since the first proposition of cell surface receptors for important roles in cardiovascular physiology and pathophysi- nucleotides [1, 2], it has become increasingly clear that, in ology, acting via P2X (ion channel) and P2Y (G protein- addition to functioning as an intracellular energy source, the coupled) receptors. In this article we consider the dual role of purines and pyrimidines ATP, adenosine diphosphate ATP in regulation of vascular tone, released as a cotransmitter (ADP), uridine triphosphate (UTP) and uridine diphosphate from sympathetic nerves or released in the vascular lumen in (UDP) can serve as important extracellular signalling response to changes in blood flow and hypoxia. Further, molecules [3, 4] acting on 13 P2X homo- and heteromul- purinergic long-term trophic and inflammatory signalling is timer ionotropic and 8 P2Y metabotropic receptor subtypes described in cell proliferation, differentiation, migration and [5, 6] (Table 1). To terminate signalling, ectonucleotidases death in angiogenesis, vascular remodelling, restenosis and are present in the circulation and on cell surfaces, rapidly atherosclerosis. The effects on haemostasis and cardiac degrading extracellular ATP into ADP, AMP and adenosine regulation is reviewed. The involvement of ATP in vascular [7, 8]. -

Prasugrel Mylan 10 Mg Film-Coated Tablets Prasugrel
Package leaflet: Information for the user Prasugrel Mylan 5 mg film-coated tablets Prasugrel Mylan 10 mg film-coated tablets prasugrel Read all of this leaflet carefully before you start taking this medicine because it contains important information for you. – Keep this leaflet. You may need to read it again. – If you have any further questions, ask your doctor or pharmacist. – This medicine has been prescribed for you only. Do not pass it on to others. It may harm them, even if their signs of illness are the same as yours. – If you get any side effects, talk to your doctor or pharmacist. This includes any possible side effects not listed in this leaflet. See section 4. What is in this leaflet 1. What Prasugrel Mylan is and what it is used for 2. What you need to know before you take Prasugrel Mylan 3. How to take Prasugrel Mylan 4. Possible side effects 5. How to store Prasugrel Mylan 6. Contents of the pack and other information 1. What Prasugrel Mylan is and what it is used for Prasugrel Mylan, which contains the active substance prasugrel, belongs to a group of medicines called antiplatelet agents. Platelets are very small cell particles that circulate in the blood. When a blood vessel is damaged, for example if it is cut, platelets clump together to help form a blood clot (thrombus). Therefore, platelets are essential to help stop bleeding. If clots form within a hardened blood vessel such as an artery they can be very dangerous as they can cut off the blood supply, causing a heart attack (myocardial infarction), stroke or death. -

Salts of Therapeutic Agents: Chemical, Physicochemical, and Biological Considerations
molecules Review Salts of Therapeutic Agents: Chemical, Physicochemical, and Biological Considerations Deepak Gupta 1, Deepak Bhatia 2 ID , Vivek Dave 3 ID , Vijaykumar Sutariya 4 and Sheeba Varghese Gupta 4,* 1 Department of Pharmaceutical Sciences, School of Pharmacy, Lake Erie College of Osteopathic Medicine, Bradenton, FL 34211, USA; [email protected] 2 ICPH Fairfax Bernard J. Dunn School of Pharmacy, Shenandoah University, Fairfax, VA 22031, USA; [email protected] 3 Wegmans School of Pharmacy, St. John Fisher College, Rochester, NY 14618, USA; [email protected] 4 Department of Pharmaceutical Sciences, USF College of Pharmacy, Tampa, FL 33612, USA; [email protected] * Correspondence: [email protected]; Tel.: +01-813-974-2635 Academic Editor: Peter Wipf Received: 7 June 2018; Accepted: 13 July 2018; Published: 14 July 2018 Abstract: The physicochemical and biological properties of active pharmaceutical ingredients (APIs) are greatly affected by their salt forms. The choice of a particular salt formulation is based on numerous factors such as API chemistry, intended dosage form, pharmacokinetics, and pharmacodynamics. The appropriate salt can improve the overall therapeutic and pharmaceutical effects of an API. However, the incorrect salt form can have the opposite effect, and can be quite detrimental for overall drug development. This review summarizes several criteria for choosing the appropriate salt forms, along with the effects of salt forms on the pharmaceutical properties of APIs. In addition to a comprehensive review of the selection criteria, this review also gives a brief historic perspective of the salt selection processes. Keywords: chemistry; salt; water solubility; routes of administration; physicochemical; stability; degradation 1. -

A Comparative Study of Molecular Structure, Pka, Lipophilicity, Solubility, Absorption and Polar Surface Area of Some Antiplatelet Drugs
International Journal of Molecular Sciences Article A Comparative Study of Molecular Structure, pKa, Lipophilicity, Solubility, Absorption and Polar Surface Area of Some Antiplatelet Drugs Milan Remko 1,*, Anna Remková 2 and Ria Broer 3 1 Department of Pharmaceutical Chemistry, Faculty of Pharmacy, Comenius University in Bratislava, Odbojarov 10, SK-832 32 Bratislava, Slovakia 2 Department of Internal Medicine, Faculty of Medicine, Slovak Medical University, Limbová 12, SK–833 03 Bratislava, Slovakia; [email protected] 3 Department of Theoretical Chemistry, Zernike Institute for Advanced Materials, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands; [email protected] * Correspondence: [email protected]; Tel.: +421-2-5011-7291 Academic Editor: Michael Henein Received: 18 February 2016; Accepted: 11 March 2016; Published: 19 March 2016 Abstract: Theoretical chemistry methods have been used to study the molecular properties of antiplatelet agents (ticlopidine, clopidogrel, prasugrel, elinogrel, ticagrelor and cangrelor) and several thiol-containing active metabolites. The geometries and energies of most stable conformers of these drugs have been computed at the Becke3LYP/6-311++G(d,p) level of density functional theory. Computed dissociation constants show that the active metabolites of prodrugs (ticlopidine, clopidogrel and prasugrel) and drugs elinogrel and cangrelor are completely ionized at pH 7.4. Both ticagrelor and its active metabolite are present at pH = 7.4 in neutral undissociated form. The thienopyridine prodrugs ticlopidine, clopidogrel and prasugrel are lipophilic and insoluble in water. Their lipophilicity is very high (about 2.5–3.5 logP values). The polar surface area, with regard to the structurally-heterogeneous character of these antiplatelet drugs, is from very large interval of values of 3–255 Å2. -

Salts of Therapeutic Agents: Chemical, Physicochemical and Biological
Preprints (www.preprints.org) | NOT PEER-REVIEWED | Posted: 16 April 2018 doi:10.20944/preprints201804.0187.v1 1 Review 2 Salts of Therapeutic Agents: Chemical, 3 Physicochemical and Biological Considerations 4 Deepak Gupta 1, Deepak Bhatia 2, Vivek Dave3, Vijaykumar Sutariya 4 and Sheeba Varghese 5 Gupta4* 6 1 Lake Eerie College of Osteopathic Medicine , School of Pharmacy, Pharmaceutical Sciences , Bradenton , FL , USA; 7 email: [email protected] 8 2 Shenandoah University - ICPH Fairfax Bernard J. Dunn School of Pharmacy, Fairfax, VA 22031USA; email: 9 [email protected] 10 3 Wegmans School of Pharmacy, St. John Fisher College, Rochester, NY, USA; email: [email protected] 11 4 Department of Pharmaceutical Sciences, USF College of Pharmacy, Tampa, FL, USA; email: 12 [email protected] 13 * Correspondence: [email protected]; Tel.: +01-813-974-2635 14 15 Abstract: Choice of the salts of therapeutic agents or active pharmaceutical ingredients (API) is 16 based on the physicochemical properties of API and the dosage form considerations. The 17 appropriate salt can have positive effect on overall therapeutic and pharmaceutical effects of API. 18 However, the incorrect salt form can negatively affect the overall pharmaceutical outcomes of the 19 API. This review addresses various criteria for choosing appropriate salt form along with the effect 20 of salt forms on API’s pharmaceutical properties. In addition to comprehensive review of the 21 criteria, this review also gives a brief historic perspective of the salt selection process. 22 23 Keywords: Chemistry, salt, water solubility, routes of administration, physicochemical, stability, 24 degradation 25 1. Introduction 26 Salt of an Active Pharmaceutical Ingredient (API) often formed to achieve desirable formulation 27 properties. -

New Drugs: T-Score for Transparency
New drugs: T-score for transparency Access to information about drugs is essential for the quality For 2011, the T-score has been revised to include the AusPAR. use of medicines. Pharmaceutical companies and regulatory agencies, such as the Therapeutic Goods Administration The revised T-scores will be as follows: (TGA), hold large quantities of information about individual drugs, but do not always share this information. To encourage T T T manufacturer provided complete clinical evaluation transparency, Australian Prescriber rates companies' willingness to provide clinical information about new drugs. T T manufacturer provided additional useful Table 1 shows how the companies have performed between information January 2009 and December 2010. The TGA is now publishing Australian Public Assessment T manufacturer provided the AusPAR and/or the Reports (AusPARs) for prescription medicines. While the product information Editorial Executive Committee welcomes this move to greater transparency, it will still ask companies to provide the clinical T manufacturer declined to supply data evaluations for their new products. While there are similarities, the AusPAR may not include all the details found in the X manufacturer did not respond to request for data regulator's clinical evaluation. Table 1 Pharmaceutical company responses to requests for clinical evaluation data for drugs marketed Jan 2009 – Dec 2010 Company Drug T T T manufacturer provided clinical evaluation Amgen denosumab Ferring degarelix Pfizer anidulafungin eletriptan PharmaLink cilostazol Phebra caffeine citrate Roche methoxy polyethylene glycol-epoetin beta tocilizumab Sanofi Pasteur H5N1 influenza vaccine Shire icatibant Wyeth methylnaltrexone T T manufacturer provided additional useful information Abbott omega-3-acid ethyl esters Celgene azacitidine Commercial Eyes melatonin CSL H1N1 influenza vaccine Japanese encephalitis vaccine Eli Lilly prasugrel Table continued.. -

Cardioprotection by Very Mild Hypothermia in Mice
67 Brief Report Cardioprotection by very mild hypothermia in mice Betül Knoop1,2#, David Naguib1,2#, Lisa Dannenberg1,2, Carolin Helten1,2, Saif Zako1,2, Christian Jung1,2, Bodo Levkau3, Maria Grandoch4, Malte Kelm1,2, Tobias Zeus1,2, Amin Polzin1,2 1Department of Cardiology, Pulmonology, and Vascular Medicine, Medical Faculty of the Heinrich Heine University Düsseldorf, Düsseldorf, Germany; 2Cardiovascular Research Institute Düsseldorf (CARID), Düsseldorf, Germany; 3Institute of Pathophysiology, West German Heart and Vascular Center, University Hospital Essen, University of Duisburg-Essen, Essen, Germany; 4Institute for Pharmacology and Clinical Pharmacology, Heinrich Heine University Düsseldorf, Düsseldorf, Germany #These authors contributed equally to this work. Correspondence to: Dr. med. Amin Polzin, PD. Klinik für Kardiologie, Pneumologie und Angiologie, Moorenstrasse 5, 40225 Düsseldorf, Germany. Email: [email protected]. Abstract: Target temperature management is recommended in post-resuscitation care. Additionally, hypothermia is a promising option in adjunctive therapy of acute myocardial infarction (MI). However, first in men data are contradicting. There are still many open questions to identify the optimal regimen and target temperature. In this study, we aimed to investigate the effect of very mild hypothermia on infarct size (IS) in mice. Mice underwent cardiac ischemia by temporary occlusion of the left anterior descending (LAD) artery under conditions of very mild hypothermia (34–36 ). Hypothermia was reached within the first 5 minutes of ischemia (temperature: 34.6±0.5 vs. 36.8±1.1 , P=0.035). Very mild hypothermia reduced IS ℃ in mice undergoing 30 minutes ischemia [IS/area at risk (AAR): 45±12% vs. 22±4%, P=0.018] as well as mice ℃ undergoing 60 minutes ischemia [IS/AAR: 67±7% vs. -
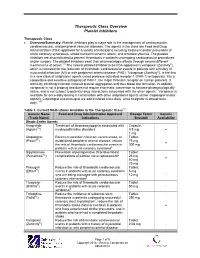
Therapeutic Class Overview Platelet Inhibitors
Therapeutic Class Overview Platelet Inhibitors Therapeutic Class • Overview/Summary: Platelet inhibitors play a major role in the management of cardiovascular, cerebrovascular, and peripheral vascular diseases. The agents in the class are Food and Drug Administration (FDA)-approved for a variety of indications including treatment and/or prevention of acute coronary syndromes, stroke/transient ischemic attack, and thrombocythemia. The platelet inhibitors are also indicated to prevent thrombosis in patients undergoing cardiovascular procedures and/or surgery. The platelet inhibitors exert their pharmacologic effects through several different mechanisms of action.1-8 The newest platelet inhibitor to be FDA-approved is vorapaxar (Zontivity®), which is indicated for the reduction of thrombotic cardiovascular events in patients with a history of myocardial infarction (MI) or with peripheral arterial disease (PAD).7 Vorapaxar (Zontivity®), is the first in a new class of antiplatelet agents called protease-activated receptor-1 (PAR-1) antagonists. It is a competitive and selective antagonist of PAR-1, the major thrombin receptor on human platelets. It works by inhibiting thrombin-induced platelet aggregation and thus blood clot formation. In addition, vorapaxar is not a prodrug and does not require enzymatic conversion to become pharmacologically active, and is not subject to potential drug interactions associated with the other agents.7 Vorapaxar is available for once-daily dosing in combination with other antiplatelet agents (either clopidogrel -

Platelet-Inhibiting Drugs: a Hematologist’S Perspective
Platelet-Inhibiting Drugs: A Hematologist’s Perspective A. Koneti Rao, M.D., Sol Sherry Professor of Medicine Chief, Hematology Section Co-Director, Sol Sherry Thrombosis Research Center Temple University School of Medicine 2012 Chile Akkerman JW, Bouma BN, Sixma JJ. Atlas of Hemostasis, 1979. STRATEGIES FOR ANTITHROMBOTIC THERAPY VASCULAR INJURY RISK FACTOR REDUCTION PLATELET ADHERANCE PLATELET AGGREGATION INHIBITORS COAGULATION ACTIVATION THROMBIN GENERATION ANTICOAGULANTS FIBRIN FORMATION PLASMIN GENERATION THROMBOLYTICS FIBRINOLYSIS AKR/2003 Platelet Responses to Activation Membrane Shape Change Aggregation Dense AGONIST Receptor Granule Secretion Thromboxane Alpha Granule Production TxA2 Acid Hydrolase ADP / Serotonin Rao/2010 GPIIb/IIIa GPIIb/IIIa Fibrinogen GPIIb/IIIa GPVI GPIa/IIa a5b1 GPIb/V/IX GPIV Damaged vWF Fibronectin Endothelium Collagen AKR/2004 Platelet-Inhibiting Drugs Platelet Inhibiting Drugs Aspirin Sulfinpyrazone Dipyridamole P2Y12 Antagonists • Thienopyridines - Ticlopidine, Clopidogrel, Prasugrel (Effient) • Non-Thienopyridine - Ticagrelor (Brilinta) GPIIb/IIIa Inhibitors Abciximab (c7E3 Fab, ReoPro) Eptifibatide (Integrilin) Tirofiban (Aggrastat) Cilostazol (Pletal) AKR-2012 Cyclooxygenase Arachidonic Acid Ser OH 529 COOH OCOCH 3 PGG2, PGH2 Aspirin Cyclooxygenase Thromboxane A2 Ser OCOCH 3 AKR/09 529 GPIIb-IIIa cAMP Fibrinogen ADP (P2Y12) Gi AC Aggregation Ticlopidine P ATP Pleckstrin Clopidogrel GP IIb/IIIa Prasugrel PKC Antagonists TxA2 ADP Gq Secretion (P2Y1) DG TS Gq PGG2/PGH2 Thrombin PLC PIP2 CO Aspirin -

Practical Guidance for the Evaluation and Management of Drug Hypersensitivity: Specific Drugs
Specific Drugs Practical Guidance for the Evaluation and Management of Drug Hypersensitivity: Specific Drugs Chief Editors: Ana Dioun Broyles, MD, Aleena Banerji, MD, and Mariana Castells, MD, PhD Ana Dioun Broyles, MDa, Aleena Banerji, MDb, Sara Barmettler, MDc, Catherine M. Biggs, MDd, Kimberly Blumenthal, MDe, Patrick J. Brennan, MD, PhDf, Rebecca G. Breslow, MDg, Knut Brockow, MDh, Kathleen M. Buchheit, MDi, Katherine N. Cahill, MDj, Josefina Cernadas, MD, iPhDk, Anca Mirela Chiriac, MDl, Elena Crestani, MD, MSm, Pascal Demoly, MD, PhDn, Pascale Dewachter, MD, PhDo, Meredith Dilley, MDp, Jocelyn R. Farmer, MD, PhDq, Dinah Foer, MDr, Ari J. Fried, MDs, Sarah L. Garon, MDt, Matthew P. Giannetti, MDu, David L. Hepner, MD, MPHv, David I. Hong, MDw, Joyce T. Hsu, MDx, Parul H. Kothari, MDy, Timothy Kyin, MDz, Timothy Lax, MDaa, Min Jung Lee, MDbb, Kathleen Lee-Sarwar, MD, MScc, Anne Liu, MDdd, Stephanie Logsdon, MDee, Margee Louisias, MD, MPHff, Andrew MacGinnitie, MD, PhDgg, Michelle Maciag, MDhh, Samantha Minnicozzi, MDii, Allison E. Norton, MDjj, Iris M. Otani, MDkk, Miguel Park, MDll, Sarita Patil, MDmm, Elizabeth J. Phillips, MDnn, Matthieu Picard, MDoo, Craig D. Platt, MD, PhDpp, Rima Rachid, MDqq, Tito Rodriguez, MDrr, Antonino Romano, MDss, Cosby A. Stone, Jr., MD, MPHtt, Maria Jose Torres, MD, PhDuu, Miriam Verdú,MDvv, Alberta L. Wang, MDww, Paige Wickner, MDxx, Anna R. Wolfson, MDyy, Johnson T. Wong, MDzz, Christina Yee, MD, PhDaaa, Joseph Zhou, MD, PhDbbb, and Mariana Castells, MD, PhDccc Boston, Mass; Vancouver and Montreal, -

Safety and Efficacy of Prasugrel with Endovascular Treatment for Unruptured Cerebral Aneurysm
Online July 5, 2017 Journal of Neuroendovascular Therapy 2017; 11: 553–557 DOI: 10.5797/jnet.oa.2017-0047 Safety and Efficacy of Prasugrel with Endovascular Treatment for Unruptured Cerebral Aneurysm Koichi Arimura, Hirotoshi Imamura, Chiaki Sakai, Shoichi Tani, Hidemitsu Adachi, Takayuki Funatsu, Mikiya Beppu, Noriyoshi Takebe, Keita Suzuki, Tomohiro Okuda, Yuichi Matsui, Yasunori Yoshida, Syuhei Kawabata, and Nobuyuki Sakai Objective: Recently, dual-antiplatelet therapy (DAPT) including clopidogrel (CLP) with endovascular treatment for unruptured cerebral aneurysm has been widely accepted. However, patients who are poor metabolizers of CLP (CLP- PMs) are more frequent in East Asians than in Caucasians, and an adequate antiplatelet effect may not be achieved with a normal dose in such patients. Prasugrel, which is a novel thienopyridine antiplatelet drug that is less likely to be poorly metabolized than CLP, is used widely for percutaneous coronary intervention, but its efficacy and safety with neuroendovascular treatment have not been elucidated. From this point of view, the purpose of this study was to elucidate the safety and efficacy of prasugrel with endovascular treatment for unruptured cerebral aneurysm. Methods: We investigated 108 consecutive patients with an unruptured cerebral aneurysm who underwent endovascular treatment from March 2015 to January 2016 in our hospital. All patients received DAPT with 100 mg aspirin and 75 mg CLP daily, and antiplatelet function was evaluated by VerifyNow (Accumetrics, San Diego, CA, USA). In patients with P2Y12 reaction units (PRU) over 230, prasugrel was administered with a loading dose of 20 mg and a maintenance dose of 3.75 mg daily. Results: Prasugrel was administered to 12 patients in our series.