Review of Chemical Processes for the Synthesis of Sodium Borohydride
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Living up to Life Special Stain Kit Modified Grocott's Methenamine
Living up to Life Special Stain Kit Modified Grocott’s Methenamine Silver Stain Catalog No: 38016SS12 Intended Use 16. Rinse slides briefly in deionized water. For In Vitro Diagnostic Use. For Laboratory Use. 17. Dehydrate slides in three changes of absolute alcohol. The reagents in this kit are intended for “In Vitro“ use only. The Modified Grocott’s Methenamine 18. Clear slides in two changes of xylene and mount in a xylene miscible medium. Silver Stain when used with appropriate histological protocols may be used for the demonstration of defined fungi and infectious agents such as Aspergillus sp., Pneumocystis Staining Protocol (Microwave) carinii and Cryptococcus neoformans in formalin fixed, paraffin embedded tissue sections. Exercise caution when using the microwave oven to heat any solution or reagent. The microwave must be properly ventilated to prevent the accumulation of fumes in the laboratory. Probable Mode of Action Microwave transparent Coplin jars and caps should be used during the staining process. The The mechanism of action of Modified Grocott’s Methenamine Silver Stain is based upon the caps should be loosely attached to prevent spills. Caps with ventilation holes also may be used. capacity of aldehyde groups to reduce cationic silver (Ag+) to metallic silver. Chromic acid is All microwave ovens should be used in accordance with the manufacturer’s instructions. The used to generate aldehyde groups by the oxidation of 1-2 glycol groups within polysaccharide procedures described here were performed using an Energy Beam Sciences H2250 laboratory rich tissue components, e.g. glycogen, mucin, reticulin and fungal cell walls. When cationic microwave. -

Aldrich FT-IR Collection Edition I Library
Aldrich FT-IR Collection Edition I Library Library Listing – 10,505 spectra This library is the original FT-IR spectral collection from Aldrich. It includes a wide variety of pure chemical compounds found in the Aldrich Handbook of Fine Chemicals. The Aldrich Collection of FT-IR Spectra Edition I library contains spectra of 10,505 pure compounds and is a subset of the Aldrich Collection of FT-IR Spectra Edition II library. All spectra were acquired by Sigma-Aldrich Co. and were processed by Thermo Fisher Scientific. Eight smaller Aldrich Material Specific Sub-Libraries are also available. Aldrich FT-IR Collection Edition I Index Compound Name Index Compound Name 3515 ((1R)-(ENDO,ANTI))-(+)-3- 928 (+)-LIMONENE OXIDE, 97%, BROMOCAMPHOR-8- SULFONIC MIXTURE OF CIS AND TRANS ACID, AMMONIUM SALT 209 (+)-LONGIFOLENE, 98+% 1708 ((1R)-ENDO)-(+)-3- 2283 (+)-MURAMIC ACID HYDRATE, BROMOCAMPHOR, 98% 98% 3516 ((1S)-(ENDO,ANTI))-(-)-3- 2966 (+)-N,N'- BROMOCAMPHOR-8- SULFONIC DIALLYLTARTARDIAMIDE, 99+% ACID, AMMONIUM SALT 2976 (+)-N-ACETYLMURAMIC ACID, 644 ((1S)-ENDO)-(-)-BORNEOL, 99% 97% 9587 (+)-11ALPHA-HYDROXY-17ALPHA- 965 (+)-NOE-LACTOL DIMER, 99+% METHYLTESTOSTERONE 5127 (+)-P-BROMOTETRAMISOLE 9590 (+)-11ALPHA- OXALATE, 99% HYDROXYPROGESTERONE, 95% 661 (+)-P-MENTH-1-EN-9-OL, 97%, 9588 (+)-17-METHYLTESTOSTERONE, MIXTURE OF ISOMERS 99% 730 (+)-PERSEITOL 8681 (+)-2'-DEOXYURIDINE, 99+% 7913 (+)-PILOCARPINE 7591 (+)-2,3-O-ISOPROPYLIDENE-2,3- HYDROCHLORIDE, 99% DIHYDROXY- 1,4- 5844 (+)-RUTIN HYDRATE, 95% BIS(DIPHENYLPHOSPHINO)BUT 9571 (+)-STIGMASTANOL -

Herbert Charles Brown, a Biographical Memoir
NATIONAL ACADEMY OF SCIENCES H E R B E R T Ch ARLES BROWN 1 9 1 2 — 2 0 0 4 A Biographical Memoir by E I-I CH I N EGIS HI Any opinions expressed in this memoir are those of the author and do not necessarily reflect the views of the National Academy of Sciences. Biographical Memoir COPYRIGHT 2008 NATIONAL ACADEMY OF SCIENCES WASHINGTON, D.C. Photograph Credit Here. HERBERT CHARLES BROWN May 22, 1912–December 19, 2004 BY EI -ICH I NEGISHI ERBERT CHARLES BROWN, R. B. Wetherill Research Profes- Hsor Emeritus of Purdue University and one of the truly pioneering giants in the field of organic-organometallic chemistry, died of a heart attack on December 19, 2004, at age 92. As it so happened, this author visited him at his home to discuss with him an urgent chemistry-related matter only about 10 hours before his death. For his age he appeared well, showing no sign of his sudden death the next morn- ing. His wife, Sarah Baylen Brown, 89, followed him on May 29, 2005. They were survived by their only child, Charles A. Brown of Hitachi Ltd. and his family. H. C. Brown shared the Nobel Prize in Chemistry in 1979 with G. Wittig of Heidelberg, Germany. Their pioneering explorations of boron chemistry and phosphorus chemistry, respectively, were recognized. Aside from several biochemists, including V. du Vigneaud in 1955, H. C. Brown was only the second American organic chemist to win a Nobel Prize behind R. B. Woodward, in 1965. His several most significant contribu- tions in the area of boron chemistry include (1) codiscovery of sodium borohyride (1972[1], pp. -

Suplementary Information
Supplementary Material (ESI) for Polymer Chemistry This journal is (c) The Royal Society of Chemistry 2011 SUPLEMENTARY INFORMATION Shedding the hydrophobic mantel of polymersomes René P. Brinkhuis, Taco R. Visser, Floris P. J. T. Rutjes, Jan C.M. van Hest* Radboud University Nijmegen,, Institute for Molecules and Materials, Heyendaalseweg 135, 6525 AJ Nijmegen, The Netherlands; E-mail: [email protected]; [email protected] Contents: Experimental General note Materials Instrumentation α- methoxy ω-methyl ester poly(ethylene glycol) α- methoxy ω-hydrazide poly(ethylene glycol) α- azido ω-methoxy poly(ethylene glycol) Aldehyde terminated polybutadiene Alkyne terminated polybutadiene Polybutadiene-Hz-poly(ethylene glycol) Polybutadiene-b-poly(ethylene glycol) Vesicle preparation Stability studies Notes and References Figures Figure 1: GPC traces of polymer 1 and its constituents. Figure 2: GPC traces of polymer 2 and its constituents. Figure 3: GPC traces of polymer 3 and its constituents. Figure 4: GPC traces of polymer 4 and its constituents. Supplementary Material (ESI) for Polymer Chemistry This journal is (c) The Royal Society of Chemistry 2011 Experimental section General note: All reactions are described for the lower molecular weight analogue, polyethylene glycol 1000, indicated with an A. The procedures for the polyethylene glycol 2000 analogues, indicated with B, are the same, starting with equimolar amounts. Materials: Sec-butyllithium (ALDRICH 1.4M in hexane), 1,3 butadiene (SIGMA ALDRICH, 99+%), 3- bromo-1-(trimethylsilyl-1-propyne) (ALDRICH, 98%), tetrabutylammonium fluoride (TBAF) (ALDRICH, 1.0M in THF), polyethylene glycol 1000 monomethyl ether (FLUKA), polyethylene glycol 2000 monomethyl ether (FLUKA), methanesulfonyl chloride (MsCl) (JANSSEN CHIMICA, 99%), sodium azide (ACROS ORGANICS, 99% extra pure), sodium hydride (ALDRICH, 60% dispersion in mineral oil), copper bromide (CuBr) (FLUKA, >98%), N,N,N’,N’,N”-penta dimethyldiethylenetriamine (PMDETA) (Aldrich,99%), hydrazine (ALDRICH, 1M in THF) were used as received. -

Scientific Committee on Consumer Safety SCCS
SCCS/1249/09 Revision of 28 September 2010 Scientific Committee on Consumer Safety SCCS OPINION ON Boron compounds The SCCS adopted this opinion at its 7th plenary meeting of 22 June 2010 SCCS/1249/09 Opinion on boron compounds ___________________________________________________________________________________________ About the Scientific Committees Three independent non-food Scientific Committees provide the Commission with the scientific advice it needs when preparing policy and proposals relating to consumer safety, public health and the environment. The Committees also draw the Commission's attention to the new or emerging problems which may pose an actual or potential threat. They are: the Scientific Committee on Consumer Safety (SCCS), the Scientific Committee on Health and Environmental Risks (SCHER) and the Scientific Committee on Emerging and Newly Identified Health Risks (SCENIHR) and are made up of external experts. In addition, the Commission relies upon the work of the European Food Safety Authority (EFSA), the European Medicines Evaluation Agency (EMA), the European Centre for Disease prevention and Control (ECDC) and the European Chemicals Agency (ECHA). SCCS The Committee shall provide opinions on questions concerning all types of health and safety risks (notably chemical, biological, mechanical and other physical risks) of non-food consumer products (for example: cosmetic products and their ingredients, toys, textiles, clothing, personal care and household products such as detergents, etc.) and services (for example: tattooing, artificial sun tanning, etc.). Scientific Committee members Jürgen Angerer, Ulrike Bernauer, Claire Chambers, Qasim Chaudhry, Gisela Degen, Gerhard Eisenbrand, Thomas Platzek, Suresh Chandra Rastogi, Vera Rogiers, Christophe Rousselle, Tore Sanner, Kai Savolainen, Jacqueline Van Engelen, Maria Pilar Vinardell, Rosemary Waring, Ian R. -

Sodium Borohydride
Version 6.6 SAFETY DATA SHEET Revision Date 01/21/2021 Print Date 09/25/2021 SECTION 1: Identification of the substance/mixture and of the company/undertaking 1.1 Product identifiers Product name : Sodium borohydride Product Number : 452882 Brand : Aldrich CAS-No. : 16940-66-2 1.2 Relevant identified uses of the substance or mixture and uses advised against Identified uses : Laboratory chemicals, Synthesis of substances 1.3 Details of the supplier of the safety data sheet Company : Sigma-Aldrich Inc. 3050 SPRUCE ST ST. LOUIS MO 63103 UNITED STATES Telephone : +1 314 771-5765 Fax : +1 800 325-5052 1.4 Emergency telephone Emergency Phone # : 800-424-9300 CHEMTREC (USA) +1-703- 527-3887 CHEMTREC (International) 24 Hours/day; 7 Days/week SECTION 2: Hazards identification 2.1 Classification of the substance or mixture GHS Classification in accordance with 29 CFR 1910 (OSHA HCS) Chemicals which, in contact with water, emit flammable gases (Category 1), H260 Acute toxicity, Oral (Category 3), H301 Skin corrosion (Category 1B), H314 Serious eye damage (Category 1), H318 Reproductive toxicity (Category 1B), H360 For the full text of the H-Statements mentioned in this Section, see Section 16. 2.2 GHS Label elements, including precautionary statements Pictogram Signal word Danger Aldrich - 452882 Page 1 of 9 The life science business of Merck KGaA, Darmstadt, Germany operates as MilliporeSigma in the US and Canada Hazard statement(s) H260 In contact with water releases flammable gases which may ignite spontaneously. H301 Toxic if swallowed. H314 Causes severe skin burns and eye damage. H360 May damage fertility or the unborn child. -
BORON Boron Compounds Are Used in the Manufacture of Glass, Soaps and Detergents and As Flame Retardants
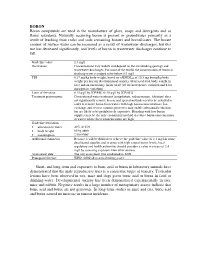
BORON Boron compounds are used in the manufacture of glass, soaps and detergents and as flame retardants. Naturally occurring boron is present in groundwater primarily as a result of leaching from rocks and soils containing borates and borosilicates. The borate content of surface water can be increased as a result of wastewater discharges, but this use has decreased significantly, and levels of boron in wastewater discharges continue to fall. Guideline value 2.4 mg/l Occurrence Concentrations vary widely and depend on the surrounding geology and wastewater discharges. For most of the world, the concentration of boron in drinking-water is judged to be below 0.5 mg/l. TDI 0.17 mg/kg body weight, based on a BMDL 05 of 10.3 mg boron/kg body weight per day for developmental toxicity (decreased fetal body weight in rats) and an uncertainty factor of 60 (10 for interspecies variation and 6 for intraspecies variation) Limit of detection 0.15 µg/l by ICP/MS; 6–10 µg/l by ICP/AES Treatment performance Conventional water treatment (coagulation, sedimentation, filtration) does not significantly remove boron, and special methods need to be installed in order to remove boron from waters with high boron concentrations. Ion exchange and reverse osmosis processes may enable substantial reduction but are likely to be prohibitively expensive. Blending with low-boron supplies may be the only economical method to reduce boron concentrations in waters where these concentrations are high. Guideline derivation • allocation to water 40% of TDI • body weight 60 kg adult • consumption 2 litres/day Additional comments Because it will be difficult to achieve the guideline value of 2.4 mg/l in some desalinated supplies and in areas with high natural boron levels, local regulatory and health authorities should consider a value in excess of 2.4 mg/l by assessing exposure from other sources. -

Part I. the Total Synthesis Of
AN ABSTRACT OF THE THESIS OF Lester Percy Joseph Burton forthe degree of Doctor of Philosophy in Chemistry presentedon March 20, 1981. Title: Part 1 - The Total Synthesis of (±)-Cinnamodialand Related Drimane Sesquiterpenes Part 2 - Photochemical Activation ofthe Carboxyl Group Via NAcy1-2-thionothiazolidines Abstract approved: Redacted for privacy DT. James D. White Part I A total synthesis of the insect antifeedant(±)-cinnamodial ( ) and of the related drimanesesquiterpenes (±)-isodrimenin (67) and (±)-fragrolide (72)are described from the diene diester 49. Hydro- boration of 49 provided the C-6oxygenation and the trans ring junction in the form of alcohol 61. To confirm the stereoselectivity of the hydroboration, 61 was convertedto both (t)-isodrimenin (67) and (±)-fragrolide (72) in 3 steps. A diisobutylaluminum hydride reduction of 61 followed by a pyridiniumchlorochromate oxidation and treatment with lead tetraacetate yielded the dihydrodiacetoxyfuran102. The base induced elimination of acetic acid preceded theepoxidation and provided 106 which contains the desired hydroxy dialdehydefunctionality of cinnamodial in a protected form. The epoxide 106 was opened with methanol to yield the acetal 112. Reduction, hydrolysis and acetylation of 112 yielded (t)- cinnamodial in 19% overall yield. Part II - Various N- acyl- 2- thionothiazolidineswere prepared and photo- lysed in the presence of ethanol to provide the corresponding ethyl esters. The photochemical activation of the carboxyl function via these derivatives appears, for practical purposes, to be restricted tocases where a-keto hydrogen abstraction and subsequent ketene formation is favored by acyl substitution. Part 1 The Total Synthesis of (±)-Cinnamodial and Related Drimane Sesquiterpenes. Part 2 Photochemical Activation of the Carboxyl Group via N-Acy1-2-thionothiazolidines. -

Safety Data Sheet
SAFETY DATA SHEET According to JIS Z 7253:2019 Revision Date 18-Aug-2020 Version 3.02 Section 1: PRODUCT AND COMPANY IDENTIFICATION Product name Sodium Metaborate Tetrahydrate Product code 198-02395 Manufacturer FUJIFILM Wako Pure Chemical Corporation 1-2 Doshomachi 3-Chome Chuo-ku, Osaka 540-8605, Japan Phone: +81-6-6203-3741 Fax: +81-6-6203-5964 Supplier FUJIFILM Wako Pure Chemical Corporation 1-2 Doshomachi 3-Chome, Chuo-ku, Osaka 540-8605, Japan Phone: +81-6-6203-3741 Fax: +81-6-6203-2029 Emergency telephone number +81-6-6203-3741 / +81-3-3270-8571 Recommended uses and For research purposes restrictions on use Section 2: HAZARDS IDENTIFICATION GHS classification Classification of the substance or mixture Skin corrosion/irritation Category 2 Serious eye damage/eye irritation Category 2A Specific target organ toxicity (single exposure) Category 3 Category 3 Respiratory tract irritation Pictograms Signal word Warning Hazard statements H315 - Causes skin irritation H319 - Causes serious eye irritation H335 - May cause respiratory irritation Precautionary statements-(Prevention) • Wash face, hands and any exposed skin thoroughly after handling • Wear protective gloves/protective clothing/eye protection/face protection • Avoid breathing dust/fume/gas/mist/vapors/spray • Use only outdoors or in a well-ventilated area Precautionary statements-(Response) • IF IN EYES: Rinse cautiously with water for several minutes. Remove contact lenses, if present and easy to do. Continue rinsing. • If eye irritation persists: Get medical advice/attention. • IF ON SKIN: Wash with plenty of soap and water • If skin irritation occurs: Get medical advice/attention • Take off contaminated clothing and wash before reuse __________________________________________________________________________________________ Page 1 / 6 W01W0119-0239 JGHEEN Sodium Metaborate Tetrahydrate Revision Date 18-Aug-2020 __________________________________________________________________________________________ • IF INHALED: Remove victim to fresh air and keep at rest in a position comfortable for breathing. -

Boranes: Physical & Chemical Properties, Encyclopaedia of Occupational Health and Safety, Jeanne Mager Stellman, Editor-In
Boranes: Physical & chemical properties, Encyclopaedia of Occupational Health and Safety, Jeanne Mager Stellman, Editor-in-Chief. International Labor Organization, Geneva. 2011. Chemical Name Colour/Form Boiling Point Melting Molecular Solubility in Relative Density Relative Vapour Inflam. Flash Auto CAS-Number (°C) Point (°C) Weight Water (water=1) Vapour Pressure/ Limits Point (°C) Ignition Density (Kpa) Point (°C) (air=1) BORON polymorphic: alpha- 2550 2300 10.81 insol Amorphous, 1.56x 580 3 -5 7440-42-8 rhombohedral form, clear 2.3 g/cm ; 10 red crystals; beta- alpha-- @ 2140 °C rhombohedral form, black; rhombohedral, - alpha-tetragonal form, 2.46 g/cm3; - black, opaque crystals with alpha-- metallic luster; amorphous tetragonal, - form, black or dark brown 2.31 g/cm3; - powder; other crystal beta-rhom- forms known bohedral, - 2.35 g/cm3 BORIC ACID, DISODIUM powder or glass-like 1575 741 201.3 2.56 g/100 g 2.367 SALT plates; white, free-flowing 1330-43-4 crystals; light grey solid BORON OXIDE rhombic crystals; 1860 450 69.6 2.77 g/100 g 1.8 1303-86-2 colourless, (amorphous); semitransparent lumps or 2.46 hard, white crystals (crystalline) BORON TRIBROMIDE colourless liquid 90 -46.0 250.57 reacts 2.6431 8.6 5.3 10294-33-4 @ 18.4 °C/4 °C @ 14 °C BORON TRICHLORIDE 12.5 -107 117.16 1.35 4.03 2.99 Pa 10294-34-5 @ 12 °C/4 @ 12.4 °C BORON TRIFLUORIDE colourless gas -99.9 -126.8 67.82 reacts 3.08g/1.57 l 2.4 10 mm Hg 7637-07-2 @ 4 °C @ -141 °C. -

Cationic Oligomerization of Ethylene Oxide
Polymer Journal, Vol. 15, No. 12, pp 883-889 (1983) Cationic Oligomerization of Ethylene Oxide Shiro KOBAYASHI, Takatoshi KOBAYASHI, and Takeo SAEGUSA Department of Synthetic Chemistry, Faculty of Engineering, Kyoto University, Kyoto 606, Japan (Received July 29, 1983) ABSTRACT: The oligomerization of ethylene oxide (EO) was investigated with several cationic catalysts to find a method for producing cyclic oligomers (crown ethers). The reactions were monitored by NMR spectroscopy (1 H and 19F) and gas chromatography. The composition of oligomers was found to change during the reactions, and the production of 1,4-dioxane increased at the later stage of the reactions. This indicates that the composition of oligomers is kinetically controlled. The formation of higher cyclic oligomers was favored by the addition of tetrahydro pyran or 1,4-dioxane to the system catalyzed by an oxonium salt; the maximum yield of cyclic tetramer was 16.8%. The effect of alkali metal salts was also examined. A template effect was observed to increase the amount of cyclic oligomer production. KEY WORDS Cationic Oligomerization I Ethylene Oxide I Cyclic Oligomers I Crown Ether I Kinetically Controlled Reaction I Additive Effects of Metal Salts I Template Effect I An extensive study was recently carried out on EXPERIMENTAL the cationic. polymerization of heterocyclic mono mers which, it was found, may lead to polymers Materials containing significant amounts of linear and cyclic All reagents were distilled under nitrogen. A oligomers.1 The most thoroughly studied monomer commercial sample of EO was distilled twice. was ethylene oxide (EO). Eastham et a!. observed Tetrahydropyran (THP) and DON were dried with that the cationic polymerization of EO resulted in sodium metal and distilled. -

Electroless Nickel, Alloy, Composite and Nano Coatings - a Critical Review
Electroless nickel, alloy, composite and nano coatings - A critical review Sudagar, J., Lian, J., & Sha, W. (2013). Electroless nickel, alloy, composite and nano coatings - A critical review. Journal of Alloys and Compounds, 571, 183–204. https://doi.org/10.1016/j.jallcom.2013.03.107 Published in: Journal of Alloys and Compounds Document Version: Peer reviewed version Queen's University Belfast - Research Portal: Link to publication record in Queen's University Belfast Research Portal Publisher rights Copyright 2013 Elsevier. This manuscript is distributed under a Creative Commons Attribution-NonCommercial-NoDerivs License (https://creativecommons.org/licenses/by-nc-nd/4.0/), which permits distribution and reproduction for non-commercial purposes, provided the author and source are cited. General rights Copyright for the publications made accessible via the Queen's University Belfast Research Portal is retained by the author(s) and / or other copyright owners and it is a condition of accessing these publications that users recognise and abide by the legal requirements associated with these rights. Take down policy The Research Portal is Queen's institutional repository that provides access to Queen's research output. Every effort has been made to ensure that content in the Research Portal does not infringe any person's rights, or applicable UK laws. If you discover content in the Research Portal that you believe breaches copyright or violates any law, please contact [email protected]. Download date:02. Oct. 2021 ELECTROLESS NICKEL, ALLOY, COMPOSITE AND NANO COATINGS - A CRITICAL REVIEW Jothi Sudagar Department of Metallurgical and Materials Engineering, Indian Institute of Technology- Madras (IIT-M), Chennai - 600 036, India Jianshe Lian College of Materials Science and Engineering, Jilin University, Changchun 130025, China.