Prediction of Sulfotransferase Specificity for Risk Assessment in Drug Design
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Targeting Fibrosis in the Duchenne Muscular Dystrophy Mice Model: an Uphill Battle
bioRxiv preprint doi: https://doi.org/10.1101/2021.01.20.427485; this version posted January 21, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Title: Targeting fibrosis in the Duchenne Muscular Dystrophy mice model: an uphill battle 2 Marine Theret1#, Marcela Low1#, Lucas Rempel1, Fang Fang Li1, Lin Wei Tung1, Osvaldo 3 Contreras3,4, Chih-Kai Chang1, Andrew Wu1, Hesham Soliman1,2, Fabio M.V. Rossi1 4 1School of Biomedical Engineering and the Biomedical Research Centre, Department of Medical 5 Genetics, 2222 Health Sciences Mall, Vancouver, BC, V6T 1Z3, Canada 6 2Department of Pharmacology and Toxicology, Faculty of Pharmaceutical Sciences, Minia 7 University, Minia, Egypt 8 3Developmental and Stem Cell Biology Division, Victor Chang Cardiac Research Institute, 9 Darlinghurst, NSW, 2010, Australia 10 4Departamento de Biología Celular y Molecular and Center for Aging and Regeneration (CARE- 11 ChileUC), Facultad de Ciencias Biológicas, Pontificia Universidad Católica de Chile, 8331150 12 Santiago, Chile 13 # Denotes Co-first authorship 14 15 Keywords: drug screening, fibro/adipogenic progenitors, fibrosis, repair, skeletal muscle. 16 Correspondence to: 17 Marine Theret 18 School of Biomedical Engineering and the Biomedical Research Centre 19 University of British Columbia 20 2222 Health Sciences Mall, Vancouver, British Columbia 21 Tel: +1(604) 822 0441 fax: +1(604) 822 7815 22 Email: [email protected] 1 bioRxiv preprint doi: https://doi.org/10.1101/2021.01.20.427485; this version posted January 21, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. -

Dr. Duke's Phytochemical and Ethnobotanical Databases Chemicals Found in Glycyrrhiza Uralensis
Dr. Duke's Phytochemical and Ethnobotanical Databases Chemicals found in Glycyrrhiza uralensis Activities Count Chemical Plant Part Low PPM High PPM StdDev Refernce Citation 0 1-METHOXY-FICIFOLINOL Root 3.4 -- 0 18-ALPHA- Root -- GLYCYRRHETINIC-ACID 0 18-ALPHA-GLYCYRRHIZIN Root 200.0 -- 0 18-ALPHA-HYDROXY- Rhizome -- GLYCYRRHETATE 0 18-BETA- Root -- GLYCYRRHETINIC-ACID 0 2',4',5-TRIHYDROXY-7- Root -- METHOXY-8-ALPHA- ALPHA-DIMETHYL-ALLYL-3- ARYLCOUMARIN 0 2',4',7-TRIHYDROXY-3'- Root -- GAMMA-GAMMA- DIMETHYL-ALLYL-3- ARYLCOUMARIN 0 2,3-DIHYDRO- Root -- ISOLIQUIRITIGENIN 0 2-METHYL-7- Root Chemical Constituents of HYDROXYISOFLAVONE Oriental Herbs (3 diff. books) 0 22-BETA-ACETYL- Root -- GLABRIC-ACID 0 24-HYDROXYGLABROLIDE Root -- 0 24- Root -- HYDROXYGLYCYRRHETIC- ACID-METHYL-ESTER 0 28- Root Chemical Constituents of HYDROXYGLYCYRRHETIC- Oriental Herbs (3 diff. books) ACID 0 3-ACETYL- Root -- GLYCYRRHETIC-ACID 0 3-BETA-24-DIHYDROXY- Root -- OLEAN-11,13(18)-DIEN-30- OIC-ACID-METHYL-ESTER 0 3-BETA- Root -- FORMYLGLABROLIDE 0 3-O-METHYLGLYCYROL Root -- 0 3-OXO-GLYCYRRHETIC- Root -- ACID 0 4',7-DIHYDROXYFLAVONE Root 240.0 -- 0 4'-O-(BETA-D-APIO-D- Root 120000.0 -- FURANOSYL-(1,2)-BETA-D- GLUCOPYRANOSYL)- LIQUIRITIGENIN Activities Count Chemical Plant Part Low PPM High PPM StdDev Refernce Citation 0 5-O-METHYLGLYCYROL Root -- 0 6''-O-ACETYL-LIQUIRITIN Root -- 0 8-C-PRENYL-ERIODICTYOL Root 13.0 -- 0 APIGENIN-6,8-DI-C- Root -- GLUCOSIDE 1 APIOGLYCYRRHIZIN Root 100.0 -1.0 -- 0 APIOISOLIQUIRITIN Root -- 0 APIOLIQUIRITIN Root -- 1 ARABOGLYCYRRHIZIN Root 600.0 1.0 -- 2 ARSENIC Root 0.3 -0.19476716146558964 Chen, H.C. -

Identification of Key Transporters Mediating Uptake of Aconitum
RSC Advances View Article Online PAPER View Journal | View Issue Identification of key transporters mediating uptake of aconitum alkaloids into the liver and kidneys and Cite this: RSC Adv.,2019,9,16136 the potential mechanism of detoxification by active ingredients of liquorice Yufei He, †a Ze Wang,†b Weidang Wu,c Ying Xie,d Zihong Wei,c Xiulin Yi,c Yong Zeng,c Yazhuo Li *c and Changxiao Liu*ac Aconite as a commonly used herb has been extensively applied in the treatment of rheumatoid arthritis, as pain relief, as well as for its cardiotonic actions. Aconitum alkaloids have been shown to be the most potent ingredients in aconite, in terms of efficacy against disease, but they are also highly toxic. Apart from neurological and cardiovascular toxicity exposed, the damage to hepatocytes and nephrocytes with long-term use of aconitum alkaloids should also be carefully considered. This study attempted to investigate the critical role of uptake transporters mediating the transport of aconitum alkaloids into the Creative Commons Attribution-NonCommercial 3.0 Unported Licence. liver and the kidneys. The resulting data revealed that hOATP1B1, 1B3, hOCT1 and hOAT3 were mainly involved in the uptake of aconitum alkaloids. Additionally, the inhibitory effects of bioactive ingredients of liquorice on uptake transporters were screened and further confirmed by determining the IC50 values. The in vitro study suggested that liquorice might lower the toxicity of aconite by reducing its exposure in the liver and/or kidneys through inhibition of uptake transporters. Eventually, the in vivo study was indicative of detoxification of liquorice by decreasing the exposure of aconitine as representative compound in liver after co-administration, even though the exposure in kidney altered was less Received 16th January 2019 significant. -

Stems for Nonproprietary Drug Names
USAN STEM LIST STEM DEFINITION EXAMPLES -abine (see -arabine, -citabine) -ac anti-inflammatory agents (acetic acid derivatives) bromfenac dexpemedolac -acetam (see -racetam) -adol or analgesics (mixed opiate receptor agonists/ tazadolene -adol- antagonists) spiradolene levonantradol -adox antibacterials (quinoline dioxide derivatives) carbadox -afenone antiarrhythmics (propafenone derivatives) alprafenone diprafenonex -afil PDE5 inhibitors tadalafil -aj- antiarrhythmics (ajmaline derivatives) lorajmine -aldrate antacid aluminum salts magaldrate -algron alpha1 - and alpha2 - adrenoreceptor agonists dabuzalgron -alol combined alpha and beta blockers labetalol medroxalol -amidis antimyloidotics tafamidis -amivir (see -vir) -ampa ionotropic non-NMDA glutamate receptors (AMPA and/or KA receptors) subgroup: -ampanel antagonists becampanel -ampator modulators forampator -anib angiogenesis inhibitors pegaptanib cediranib 1 subgroup: -siranib siRNA bevasiranib -andr- androgens nandrolone -anserin serotonin 5-HT2 receptor antagonists altanserin tropanserin adatanserin -antel anthelmintics (undefined group) carbantel subgroup: -quantel 2-deoxoparaherquamide A derivatives derquantel -antrone antineoplastics; anthraquinone derivatives pixantrone -apsel P-selectin antagonists torapsel -arabine antineoplastics (arabinofuranosyl derivatives) fazarabine fludarabine aril-, -aril, -aril- antiviral (arildone derivatives) pleconaril arildone fosarilate -arit antirheumatics (lobenzarit type) lobenzarit clobuzarit -arol anticoagulants (dicumarol type) dicumarol -

Recent Advances in the Biological Importance of Rhodanine Derivatives
Chapter 2 Recent Advances in the Biological Importance of Rhodanine Derivatives Amit B. Patel and Premlata Kumari Additional information is available at the end of the chapter http://dx.doi.org/10.5772/62835 Abstract Heterocyclic compounds are an important part of the synthetic medicinal chemistry. They offer a high degree of structural variety and have proven to be widely useful as therapeutic agents. Heterocyclic compounds play an important role in the biological processes. They are widespread as natural products. Heterocyclic compounds are widely found in nature categorically in plant alkaloids, nucleic acids, anthocyanins, and fla‐ vones. They are also present as in chlorophyll and hemoglobin. Additionally, some pro‐ teins, hormones, and vitamins also contain aromatic heterocyclic system. Heterocycles have huge potential as the most promising molecules as lead structures for the design of new drugs. About one half of over 6 million compounds recorded so far in chemical ab‐ stracts are heterocyclic. The proposed book chapter entitled, Recent Advances in the Biologi‐ cal Importance of Rhodanine Derivatives gives an outline of importance and applications of the various rhodanine derivatives in medicinal chemistry from 2004 to 2014. Keywords: Rhodanine, biological activities, structure activity relationship and selectivity of rhodanine derivatives 1. Introduction Rhodanine is a five-membered heterocyclic molecule containing a thiazole nucleus with thioxo group on second carbon and carbonyl group on fourth carbon. It was first discovered in 1877 by Marceli Nencki, who named it “Rhodaninsaure.” Structural modifications of rhodanine derivatives (Figure 1) constantly result in compounds with a broad spectrum of pharmaco‐ logical activities [1, 2]. Rhodanine derivatives recently have grabbed the attention of research‐ ers because of their broad range of pharmacological activities. -

TE INI (19 ) United States (12 ) Patent Application Publication ( 10) Pub
US 20200187851A1TE INI (19 ) United States (12 ) Patent Application Publication ( 10) Pub . No .: US 2020/0187851 A1 Offenbacher et al. (43 ) Pub . Date : Jun . 18 , 2020 ( 54 ) PERIODONTAL DISEASE STRATIFICATION (52 ) U.S. CI. AND USES THEREOF CPC A61B 5/4552 (2013.01 ) ; G16H 20/10 ( 71) Applicant: The University of North Carolina at ( 2018.01) ; A61B 5/7275 ( 2013.01) ; A61B Chapel Hill , Chapel Hill , NC (US ) 5/7264 ( 2013.01 ) ( 72 ) Inventors: Steven Offenbacher, Chapel Hill , NC (US ) ; Thiago Morelli , Durham , NC ( 57 ) ABSTRACT (US ) ; Kevin Lee Moss, Graham , NC ( US ) ; James Douglas Beck , Chapel Described herein are methods of classifying periodontal Hill , NC (US ) patients and individual teeth . For example , disclosed is a method of diagnosing periodontal disease and / or risk of ( 21) Appl. No .: 16 /713,874 tooth loss in a subject that involves classifying teeth into one of 7 classes of periodontal disease. The method can include ( 22 ) Filed : Dec. 13 , 2019 the step of performing a dental examination on a patient and Related U.S. Application Data determining a periodontal profile class ( PPC ) . The method can further include the step of determining for each tooth a ( 60 ) Provisional application No.62 / 780,675 , filed on Dec. Tooth Profile Class ( TPC ) . The PPC and TPC can be used 17 , 2018 together to generate a composite risk score for an individual, which is referred to herein as the Index of Periodontal Risk Publication Classification ( IPR ) . In some embodiments , each stage of the disclosed (51 ) Int. Cl. PPC system is characterized by unique single nucleotide A61B 5/00 ( 2006.01 ) polymorphisms (SNPs ) associated with unique pathways , G16H 20/10 ( 2006.01 ) identifying unique druggable targets for each stage . -

Insecticidal and Antifungal Chemicals Produced by Plants
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Archive Ouverte en Sciences de l'Information et de la Communication Insecticidal and antifungal chemicals produced by plants: a review Isabelle Boulogne, Philippe Petit, Harry Ozier-Lafontaine, Lucienne Desfontaines, Gladys Loranger-Merciris To cite this version: Isabelle Boulogne, Philippe Petit, Harry Ozier-Lafontaine, Lucienne Desfontaines, Gladys Loranger- Merciris. Insecticidal and antifungal chemicals produced by plants: a review. Environmental Chem- istry Letters, Springer Verlag, 2012, 10 (4), pp.325 - 347. 10.1007/s10311-012-0359-1. hal-01767269 HAL Id: hal-01767269 https://hal-normandie-univ.archives-ouvertes.fr/hal-01767269 Submitted on 29 May 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Distributed under a Creative Commons Attribution - NonCommercial| 4.0 International License Version définitive du manuscrit publié dans / Final version of the manuscript published in : Environmental Chemistry Letters, 2012, n°10(4), 325-347 The final publication is available at www.springerlink.com : http://dx.doi.org/10.1007/s10311-012-0359-1 Insecticidal and antifungal chemicals produced by plants. A review Isabelle Boulogne 1,2* , Philippe Petit 3, Harry Ozier-Lafontaine 2, Lucienne Desfontaines 2, Gladys Loranger-Merciris 1,2 1 Université des Antilles et de la Guyane, UFR Sciences exactes et naturelles, Campus de Fouillole, F- 97157, Pointe-à-Pitre Cedex (Guadeloupe), France. -

SULT1E1 Inhibits Cell Proliferation and Invasion by Activating Pparγ in Breast Cancer
Journal of Cancer 2018, Vol. 9 1078 Ivyspring International Publisher Journal of Cancer 2018; 9(6): 1078-1087. doi: 10.7150/jca.23596 Research Paper SULT1E1 inhibits cell proliferation and invasion by activating PPARγ in breast cancer Yali Xu1, Xiaoyan Lin1, Jiawen Xu1, Haiyan Jing1, Yejun Qin1, Yintao Li2 1. Department of Pathology, Shandong Provincial Hospital Affiliated to Shandong University, Jinan, Shandong, P.R. China 2. Department of Medical Oncology, Shandong Cancer Hospital and Institute, Jinan, Shandong, P.R. China Corresponding author: Yejun Qin, Department of Pathology, Shandong Provincial Hospital Affiliated to Shandong University, No. 324 Jingwu Road, Jinan, Shandong, 250021, China. Tel: +86-531-68776430. E-mail: [email protected] and Yintao Li, Department of Medical Oncology, Shandong Cancer Hospital and Institute, Shandong University, No. 440 Jiefang Road, Jinan, Shandong, 250117, China. Tel: +86-531-87984777. E-mail: [email protected] © Ivyspring International Publisher. This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions. Received: 2017.10.31; Accepted: 2018.01.29; Published: 2018.02.28 Abstract Sulfotransferase family 1E member 1 (SULT1E1) is known to catalyze sulfoconjugation and play a crucial role in the deactivation of estrogen homeostasis, which is involved in tumorigenesis and the progression of breast and endometrial cancers. Our previous study has shown that the protein levels of SULT1E1 were decreased in breast cancer; however, the underlying mechanism is still poorly understood. In this study, we explored the functional and molecular mechanisms by which SULT1E1 influenced breast cancer. -
Downloaded from Survive Nursing | Survivenursing.Com V20110426
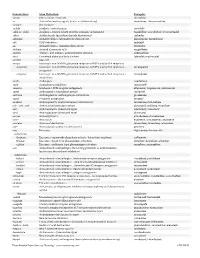
Generic Stem Stem Definition Examples -abine (see -arabine, -citabine) decitabine -ac Anti-inflammatory agents (acetic acid derivatives) bromfenac; dexpemedolac -acetam See -racetam -actide Synthetic corticotropins seractide -adol or -aldol- Analgesics (mixed opiate receptor agonists/ antagonists) tazadolene; spiradolene; levonantradol -adox Antibacterials (quinoline dioxide derivatives) carbadox -afenone Antiarrhythmics (propafenone derivatives) alprafenone; diprafenone -afil PDE5 inhibitors tadalafil -aj- Antiarrhythmics (ajmaline derivatives) lorajmine -aldrate Antacid aluminum salts magaldrate -algron Alpha1 - and alpha2 - adrenoreceptor agonists dabuzalgron -alol Combined alpha and beta blockers labetalol; medroxalol -amivir (see -vir) -ampa Ionotropic non-NMDA glutamate receptors (AMPA and/or KA receptors) -ampanel Ionotropic non-NMDA glutamate receptors (AMPA and/or KA receptors) ; becampanel antagonists -ampator Ionotropic non-NMDA glutamate receptors (AMPA and/or KA receptors) ; forampator modulators -andr- Androgens nandrolone -anib Angiogenesis inhibitors semaxanib -anserin Serotonin 5-HT2 receptor antagonists altanserin; tropanserin; adatanserin -antel Anthelmintics (undefined group) carbantel -antrone Antineoplastics; anthraquinone derivatives pixantrone -apsel P-selectin antagonists torapsel -arabine Antineoplastics (arabinofuranosyl derivatives) fazarabine; fludarabine aril-, -aril, -aril- Antiviral (arildone derivatives) pleconaril; arildone; fosarilate -arit Antirheumatics (lobenzarit type) lobenzarit; clobuzarit -arol -

Datasheet Inhibitors / Agonists / Screening Libraries a DRUG SCREENING EXPERT
Datasheet Inhibitors / Agonists / Screening Libraries A DRUG SCREENING EXPERT Product Name : Prinaberel Catalog Number : TQ0149 CAS Number : 524684-52-4 Molecular Formula : C15H10FNO3 Molecular Weight : 271.24 Description: Prinaberel (ERB-041) is an effective and selective ERβ agonist and >200-fold selective for ERβ. Storage: 2 years -80°C in solvent; 3 years -20°C powder; Solubility DMSO ≥38mg/mL(140.1 mM) ( < 1 mg/ml refers to the product slightly soluble or insoluble ) In vitro Activity Treatment with ERβ selective estrogen agonists liquiritigenin and ERB-041 reduced the ability to invade a reconstituted basement membrane and to migrate in response to the cellular stimulus [1]. Pretreatment the PMs with ERB-041 resulted in significant inhibition of LPS-induced iNOS expression and NF-kappaB activation by preventing its nuclear translocation [3]. In vivo Activity Tumor numbers and volume were reduced by 60% and 84%, respectively, in the Erb-041-treated group as compared with UVB (alone) control. This inhibition in tumorigenesis was accompanied by a decrease in proliferating cell nuclear antigen (PCNA), cyclin D1, VEGF, and CD31, and an increase in apoptosis [2]. Reference 1. Hinsche O, et al. Estrogen receptor β selective agonists reduce invasiveness of triple-negative breast cancer cells. Int J Oncol. 2015 Feb;46(2):878-84. 2. Chaudhary SC, et al. Erb-041, an estrogen receptor-β agonist, inhibits skin photocarcinogenesis in SKH-1 hairless mice by downregulating the WNT signaling pathway. Cancer Prev Res (Phila). 2014 Feb;7(2):186-98. 3. Xiu-li W, et al. ERB-041, a selective ER beta agonist, inhibits iNOS production in LPS-activated peritoneal macrophages of endometriosis via suppression of NF-kappaB activation. -

Simultaneous Determination and Pharmacokinetic Characterization
molecules Article Simultaneous Determination and Pharmacokinetic Characterization of Glycyrrhizin, Isoliquiritigenin, Liquiritigenin, and Liquiritin in Rat Plasma Following Oral Administration of Glycyrrhizae Radix Extract You Jin Han 1, Bitna Kang 2, Eun-Ju Yang 1, Min-Koo Choi 2,* and Im-Sook Song 1,* 1 College of Pharmacy and Research Institute of Pharmaceutical Sciences, Kyungpook National University, Daegu 41566, Korea; [email protected] (Y.J.H.); [email protected] (E.-J.Y.) 2 College of Pharmacy, Dankook University, Cheon-an 31116, Korea; [email protected] * Correspondence: [email protected] (I.-S.S.); [email protected] (M.-K.C.); Tel.: +82-53-950-8575 (I.-S.S.); +82-41-550-1432 (M.-K.C.) Academic Editors: In-Soo Yoon and Hyun-Jong Cho Received: 12 April 2019; Accepted: 9 May 2019; Published: 10 May 2019 Abstract: Glycyrrhizae Radix is widely used as herbal medicine and is effective against inflammation, various cancers, and digestive disorders. We aimed to develop a sensitive and simultaneous analytical method for detecting glycyrrhizin, isoliquiritigenin, liquiritigenin, and liquiritin, the four marker components of Glycyrrhizae Radix extract (GRE), in rat plasma using liquid chromatography-tandem mass spectrometry and to apply this analytical method to pharmacokinetic studies. Retention times for glycyrrhizin, isoliquiritigenin, liquiritigenin, and liquiritin were 7.8 min, 4.1 min, 3.1 min, and 2.0 min, respectively, suggesting that the four analytes were well separated without any interfering peaks around the peak elution time. The lower limit of quantitation was 2 ng/mL for glycyrrhizin and 0.2 ng/mL for isoliquiritigenin, liquiritigenin, and liquiritin; the inter- and intra-day accuracy, precision, and stability were less than 15%. -

Assembly of a Novel Biosynthetic Pathway for Production of the Plant Flavonoid Fisetin in Escherichia Coli
Downloaded from orbit.dtu.dk on: Oct 06, 2021 Assembly of a novel biosynthetic pathway for production of the plant flavonoid fisetin in Escherichia coli Stahlhut, Steen Gustav; Siedler, Solvej; Malla, Sailesh; Harrison, Scott James; Maury, Jerome; Neves, Ana Rute; Förster, Jochen Published in: Metabolic Engineering Link to article, DOI: 10.1016/j.ymben.2015.07.002 Publication date: 2015 Document Version Publisher's PDF, also known as Version of record Link back to DTU Orbit Citation (APA): Stahlhut, S. G., Siedler, S., Malla, S., Harrison, S. J., Maury, J., Neves, A. R., & Förster, J. (2015). Assembly of a novel biosynthetic pathway for production of the plant flavonoid fisetin in Escherichia coli. Metabolic Engineering, 31, 84-93. https://doi.org/10.1016/j.ymben.2015.07.002 General rights Copyright and moral rights for the publications made accessible in the public portal are retained by the authors and/or other copyright owners and it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights. Users may download and print one copy of any publication from the public portal for the purpose of private study or research. You may not further distribute the material or use it for any profit-making activity or commercial gain You may freely distribute the URL identifying the publication in the public portal If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim. Metabolic Engineering 31 (2015) 84–93 Contents lists available at ScienceDirect Metabolic Engineering journal homepage: www.elsevier.com/locate/ymben Assembly of a novel biosynthetic pathway for production of the plant flavonoid fisetin in Escherichia coli Steen G.