Actinide Separations Conference Program, May 2021
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Key Official US and IAEA Statements About Iran's Nuclear Programs
Key Official US and IAEA Statements About Iran’s Nuclear Programs Anthony H. Cordesman There is a great deal of speculation about Iran’s nuclear programs that do not list sources or reflect the views of the US intelligence community. It is worth examining what top US intelligence official have said during the last few years, and the details of the IAEA report published in November 2011 – one that clearly reflected official inputs from the US and a number of European intelligence services. U.S. Official Statements on the Iranian Nuclear and Missile Threat The annual unclassified reports to Congress by the Office of the Director of National Intelligence are a key source of US official view. Although the 2010 report by James R. Clapper has already been partly overtaken by the pace of Iran’s rapidly developing program, it still represents the most detailed unclassified estimate of Iran’s capabilities by a senior US official:1 Nuclear We continue to assess Iran is keeping open the option to develop nuclear weapons though we do not know whether Tehran eventually will decide to produce nuclear weapons. Iran continues to develop a range of capabilities that could be applied to producing nuclear weapons, if a decision is made to do so. During the reporting period, Iran continued to expand its nuclear infrastructure and continued uranium enrichment and activities related to its heavy water research reactor, despite multiple United Nations Security Council Resolutions since late 2006 calling for the suspension of those activities. Although Iran made progress in expanding its nuclear infrastructure during 2001, some obstacles slowed progress during this period. -

Depleted Uranium Hexafluoride: Waste Or Resource?
UCRGJC-120397 PREPRINT Depleted Uranium Hexafluoride: Waste or Resource? N. Schwertz J. Zoller R Rosen S. Patton C. Bradley A. Murray This paper was prepared for submittal to the Global ‘95 International Conference on Evaluation of Emerging Nuclear Fuel Cycle Systems Versailles, France September 11-14,1995 July 1995 This isa preprint of apaper intended for publication in a jaurnal orproceedings. Since changes may be made before publication, this preprint is made available with the understanding that it will not be cited or reproduced without the permiasion of the anthor. DISCLAIMER This document was prepared as an account of work sponsored by an agency of the United Stat= Government. Neither theunited States Governmentmor theuniversity of California nor any oftheir employees, makes any warranty, express or implied, or assumesanylegalliabilityorrespomibility forthe accuracy,completeness,orusefuin~ of any information, apparatus, pduct, or process disdosed, or represents that its use wouldnotinfringe privatelyowned rights. Referencehemin to anyspe&c commercial prodocis, proms, or service by trade name, trademark, manufacturer, or otherwise, does not necessarily constituteor imply its endorsement, reconunendation, or favoring by the United States Government or the University of California. The views and opinions of authors expressed herein do not necessady state or reflect those of the United States Government or the University of California, and shall not be used for adveltising or product endorsement purposes. DISCLAIMER Portions of this document may be illegible in electronic image products. Images are produced from the best available original document . DEPLETED URANIUM HEXAFLUORIDE: WASTE OR RESOURCE? N. Schwertz, J. Zoller, R. Rosen, S. Patton LAWRENCE LIVERMORE NATIONAL LABORATORY P. -

Actinide Ground-State Properties-Theoretical Predictions
Actinide Ground-State Properties Theoretical predictions John M. Wills and Olle Eriksson electron-electron correlations—the electronic energy of the ground state of or nearly fifty years, the actinides interactions among the 5f electrons and solids, molecules, and atoms as a func- defied the efforts of solid-state between them and other electrons—are tional of electron density. The DFT Ftheorists to understand their expected to affect the bonding. prescription has had such a profound properties. These metals are among Low-symmetry crystal structures, impact on basic research in both the most complex of the long-lived relativistic effects, and electron- chemistry and solid-state physics that elements, and in the solid state, they electron correlations are very difficult Walter Kohn, its main inventor, was display some of the most unusual to treat in traditional electronic- one of the recipients of the 1998 behaviors of any series in the periodic structure calculations of metals and, Nobel Prize in Chemistry. table. Very low melting temperatures, until the last decade, were outside the In general, it is not possible to apply large anisotropic thermal-expansion realm of computational ability. And DFT without some approximation. coefficients, very low symmetry crystal yet, it is essential to treat these effects But many man-years of intense research structures, many solid-to-solid phase properly in order to understand the have yielded reliable approximate transitions—the list is daunting. Where physics of the actinides. Electron- expressions for the total energy in does one begin to put together an electron correlations are important in which all terms, except for a single- understanding of these elements? determining the degree to which 5f particle kinetic-energy term, can be In the last 10 years, together with electrons are localized at lattice sites. -

Uranium Aerosols at a Nuclear Fuel Fabrication Plant: Characterization Using Scanning Electron Microscopy and Energy Dispersive X-Ray Spectroscopy
Spectrochimica Acta Part B 131 (2017) 130–137 Contents lists available at ScienceDirect Spectrochimica Acta Part B journal homepage: www.elsevier.com/locate/sab Uranium aerosols at a nuclear fuel fabrication plant: Characterization using scanning electron microscopy and energy dispersive X-ray spectroscopy E. Hansson a,b,⁎, H.B.L. Pettersson a, C. Fortin c, M. Eriksson a,d a Department of Medical and Health Sciences, Linköping University, 58185 Linköping, Sweden b Westinghouse Electric Sweden AB, Bränslegatan 1, 72136 Västerås, Sweden c Carl Zeiss SAS, 100 route de Versailles, 78160 Marly-le-Roi, France d Swedish Radiation Safety Authority, 17116 Stockholm, Sweden article info abstract Article history: Detailed aerosol knowledge is essential in numerous applications, including risk assessment in nuclear industry. Received 31 March 2016 Cascade impactor sampling of uranium aerosols in the breathing zone of nuclear operators was carried out at a Received in revised form 28 February 2017 nuclear fuel fabrication plant. Collected aerosols were evaluated using scanning electron microscopy and energy Accepted 3 March 2017 dispersive X-ray spectroscopy. Imaging revealed remarkable variations in aerosol morphology at the different Available online 6 March 2017 workshops, and a presence of very large particles (up to≅100 × 50 μm2) in the operator breathing zone. Charac- teristic X-ray analysis showed varying uranium weight percentages of aerosols and, frequently, traces of nitrogen, Keywords: fl Uranium uorine and iron. The analysis method, in combination with cascade impactor sampling, can be a powerful tool Aerosol for characterization of aerosols. The uranium aerosol source term for risk assessment in nuclear fuel fabrication Impactor appears to be highly complex. -
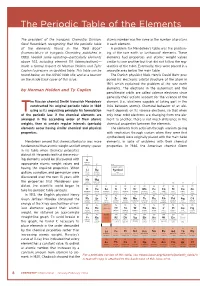
The Periodic Table of the Elements
The Periodic Table of the Elements The president of the Inorganic Chemistry Division, atomic number was the same as the number of protons Gerd Rosenblatt, recognizing that the periodic table in each element. of the elements found in the “Red Book” A problem for Mendeleev’s table was the position- (Nomenclature of Inorganic Chemistry, published in ing of the rare earth or lanthanoid* elements. These 1985) needed some updating—particularly elements elements had properties and atomic weight values above 103, including element 110 (darmstadtium)— similar to one another but that did not follow the reg- made a formal request to Norman Holden and Tyler ularities of the table. Eventually, they were placed in a Coplen to prepare an updated table. This table can be separate area below the main table. found below, on the IUPAC Web site, and as a tear-off The Danish physicist Niels Henrik David Bohr pro- on the inside back cover of this issue. posed his electronic orbital structure of the atom in 1921, which explained the problem of the rare earth by Norman Holden and Ty Coplen elements. The electrons in the outermost and the penultimate orbits are called valence electrons since generally their actions account for the valence of the he Russian chemist Dmitri Ivanovich Mendeleev element (i.e., electrons capable of taking part in the constructed his original periodic table in 1869 links between atoms). Chemical behavior of an ele- Tusing as its organizing principle his formulation ment depends on its valence electrons, so that when of the periodic law: if the chemical elements are only inner orbit electrons are changing from one ele- arranged in the ascending order of their atomic ment to another, there is not much difference in the weights, then at certain regular intervals (periods) chemical properties between the elements. -

ELECTRONEGATIVITY D Qkq F=
ELECTRONEGATIVITY The electronegativity of an atom is the attracting power that the nucleus has for it’s own outer electrons and those of it’s neighbours i.e. how badly it wants electrons. An atom’s electronegativity is determined by Coulomb’s Law, which states, “ the size of the force is proportional to the size of the charges and inversely proportional to the square of the distance between them”. In symbols it is represented as: kq q F = 1 2 d 2 where: F = force (N) k = constant (dependent on the medium through which the force is acting) e.g. air q1 = charge on an electron (C) q2 = core charge (C) = no. of protons an outer electron sees = no. of protons – no. of inner shell electrons = main Group Number d = distance of electron from the nucleus (m) Examples of how to calculate the core charge: Sodium – Atomic number 11 Electron configuration 2.8.1 Core charge = 11 (no. of p+s) – 10 (no. of inner e-s) +1 (Group i) Chlorine – Atomic number 17 Electron configuration 2.8.7 Core charge = 17 (no. of p+s) – 10 (no. of inner e-s) +7 (Group vii) J:\Sciclunm\Resources\Year 11 Chemistry\Semester1\Notes\Atoms & The Periodic Table\Electronegativity.doc Neon holds onto its own electrons with a core charge of +8, but it can’t hold any more electrons in that shell. If it was to form bonds, the electron must go into the next shell where the core charge is zero. Group viii elements do not form any compounds under normal conditions and are therefore given no electronegativity values. -

Chemistry of the Noble Gases*
CHEMISTRY OF THE NOBLE GASES* By Professor K. K. GREE~woon , :.\I.Sc., sc.D .. r".lU.C. University of N ewca.stle 1tpon Tyne The inert gases, or noble gases as they are elements were unsuccessful, and for over now more appropriately called, are a remark 60 years they epitomized chemical inertness. able group of elements. The lightest, helium, Indeed, their electron configuration, s2p6, was recognized in the gases of the sun before became known as 'the stable octet,' and this it was isolated on ea.rth as its name (i]A.tos) fotmed the basis of the fit·st electronic theory implies. The first inert gas was isolated in of valency in 1916. Despite this, many 1895 by Ramsay and Rayleigh; it was named people felt that it should be possible to induce argon (apy6s, inert) and occurs to the extent the inert gases to form compounds, and many of 0·93% in the earth's atmosphere. The of the early experiments directed to this end other gases were all isolated before the turn have recently been reviewed.l of the century and were named neon (v€ov, There were several reasons why chemists new), krypton (KpVn'TOV, hidden), xenon believed that the inert gases might form ~€vov, stmnger) and radon (radioactive chemical compounds under the correct con emanation). Though they occur much less ditions. For example, the ionization poten abundantly than argon they cannot strictly tial of xenon is actually lower than those of be called rare gases; this can be illustrated hydrogen, nitrogen, oxygen, fl uorine and by calculating the volumes occupied a.t s.t.p. -

Is Selenium a Metal, Non-Metal Or Metalloid?
Is Selenium a metal, non-metal or metalloid? Abstract Is selenium(Se) a metal, non-metal, or a metalloid? There are various public opinions circulating around it. Since a long time from now, there are a lot of voices discussing this. Even until now, there is still no consensus about it. So, in this project, we are trying to find out whether selenium is a non-metal, metal or metalloids base on its physical and chemical properties which could be studied in the secondary school combining with the other information from the internet. Principles and hypothesis Studied from the secondary school chemistry, the general properties of metals include being good conductors of heat and electricity, having high melting and boiling points. Non-metals generally have a lower melting point and boiling point than metals and they being poor conductors of heat and electricity, etc. And the physical properties of metalloids are having extremely high melting/ boiling point, and having fair electrical conductivity. On the other hand, the oxides of metal are generally basic whereas the oxide of non-metals and metalloids are generally acidic. We will define selenium’s chemical category based on the above properties. Besides, we would like to introduce the concept of displacement reaction in studying the chemical properties of the chalcogens(e.g. sulphur(S) and selenium(Se)), especially selenium such rarely mentioned element. We can assume selenium is a metal if selenium could displace a metal oxide or a metal could displace selenium (IV) oxide. If selenium is a non-metal, its oxide could be displaced by sulphur which is supposed to be more reactive than selenium in the chalcogen group. -

Periodic Table of the Elements Notes
Periodic Table of the Elements Notes Arrangement of the known elements based on atomic number and chemical and physical properties. Divided into three basic categories: Metals (left side of the table) Nonmetals (right side of the table) Metalloids (touching the zig zag line) Basic Organization by: Atomic structure Atomic number Chemical and Physical Properties Uses of the Periodic Table Useful in predicting: chemical behavior of the elements trends properties of the elements Atomic Structure Review: Atoms are made of protons, electrons, and neutrons. Elements are atoms of only one type. Elements are identified by the atomic number (# of protons in nucleus). Energy Levels Review: Electrons are arranged in a region around the nucleus called an electron cloud. Energy levels are located within the cloud. At least 1 energy level and as many as 7 energy levels exist in atoms Energy Levels & Valence Electrons Energy levels hold a specific amount of electrons: 1st level = up to 2 2nd level = up to 8 3rd level = up to 8 (first 18 elements only) The electrons in the outermost level are called valence electrons. Determine reactivity - how elements will react with others to form compounds Outermost level does not usually fill completely with electrons Using the Table to Identify Valence Electrons Elements are grouped into vertical columns because they have similar properties. These are called groups or families. Groups are numbered 1-18. Group numbers can help you determine the number of valence electrons: Group 1 has 1 valence electron. Group 2 has 2 valence electrons. Groups 3–12 are transition metals and have 1 or 2 valence electrons. -

Nuclear Iran a Glossary of Terms
Nuclear Iran A Glossary of Terms Simon Henderson and Olli Heinonen Policy Focus 121 | May 2013 Update HARVARD Kennedy School A COPUBLICATION WITH BELFER CENTER for Science and International Affairs Map: Nuclear installations in Iran. TURKMENISTAN TABRIZ Bonab Lashkar Abad TEHRAN MASHAD Karaj Marivan Parchin Fordow Arak QOM IRAQ Natanz AFGHANISTAN Isfahan Ardakan Saghand Darkhovin Yazd IRAN KUWAIT SHIRAZ Bushehr PAKISTAN Gchine BANDAR ABBAS BAHRAIN SAUDI ARABIA QATAR © 2012 The Washington Institute for Near East Policy UAE OMAN Map: Nuclear installations in Iran. Nuclear Iran A Glossary of Terms Simon Henderson and Olli Heinonen Policy Focus 121 | May 2013 Update HARVARD Kennedy School A COPUBLICATION WITH BELFER CENTER for Science and International Affairs n n n The authors extend special thanks to Mary Kalbach Horan and her editorial team at The Washington Institute. n n n All rights reserved. Printed in the United States of America. No part of this publication may be reproduced or transmitted in any form or by any means, electronic or mechanical, including photocopy, recording, or any information storage and retrieval system, without permission in writing from the publisher. © 2012, 2013 by The Washington Institute for Near East Policy and the Harvard Kennedy School’s Belfer Center for Science and International Affairs Copublished in 2012 and 2013 in the United States of America by The Washington Institute for Near East Policy, 1828 L Street NW, Suite 1050, Washington, DC 20036; and the Harvard Kennedy School’s Belfer Center for Science and International Affairs, 79 JFK St., Cambridge, MA 02138. Cover photo: Iran’s president Mahmoud Ahmadinejad visits the Natanz nuclear enrichment facility. -

The Noble Gases
INTERCHAPTER K The Noble Gases When an electric discharge is passed through a noble gas, light is emitted as electronically excited noble-gas atoms decay to lower energy levels. The tubes contain helium, neon, argon, krypton, and xenon. University Science Books, ©2011. All rights reserved. www.uscibooks.com Title General Chemistry - 4th ed Author McQuarrie/Gallogy Artist George Kelvin Figure # fig. K2 (965) Date 09/02/09 Check if revision Approved K. THE NOBLE GASES K1 2 0 Nitrogen and He Air P Mg(ClO ) NaOH 4 4 2 noble gases 4.002602 1s2 O removal H O removal CO removal 10 0 2 2 2 Ne Figure K.1 A schematic illustration of the removal of O2(g), H2O(g), and CO2(g) from air. First the oxygen is removed by allowing the air to pass over phosphorus, P (s) + 5 O (g) → P O (s). 20.1797 4 2 4 10 2s22p6 The residual air is passed through anhydrous magnesium perchlorate to remove the water vapor, Mg(ClO ) (s) + 6 H O(g) → Mg(ClO ) ∙6 H O(s), and then through sodium hydroxide to remove 18 0 4 2 2 4 2 2 the carbon dioxide, NaOH(s) + CO2(g) → NaHCO3(s). The gas that remains is primarily nitrogen Ar with about 1% noble gases. 39.948 3s23p6 36 0 The Group 18 elements—helium, K-1. The Noble Gases Were Kr neon, argon, krypton, xenon, and Not Discovered until 1893 83.798 radon—are called the noble gases 2 6 4s 4p and are noteworthy for their rela- In 1893, the English physicist Lord Rayleigh noticed 54 0 tive lack of chemical reactivity. -

Sorption of Np and Tc in Underground Waters by Uranium Oxides
Sorption of Np and Tc in Underground Waters by Uranium Oxides T.V.Kazakovskaya,a V.I. Shapovalova, N.V. Kushnira , E.V. Zakharovab, S.N.Kalmykovb O.N.Batukb, M.J.Hairec a –Russian Federal Nuclear center –All-Russia Scientific Institute of Experimental Physics; 607190, Sarov, Russia, E-mail: [email protected]; b – Institute of Physical Chemistry and Electrochemistry, Russian Academy of Sciences, Moscow, Russia, c – Oak Ridge National Laboratory, Oak Ridge, Tennessee. Abstract –The production of nuclear fuels results in the accumulation of large quantities of depleted uranium (DU) in the form of uranium hexafluoride (UF6), which is converted to uranium oxides. Depleted uranium dioxide (DUO2) can be used as a component of radiation shielding and as an absorbent for migrating radionuclides (especially 237Np and 99Tc). These may emerge from casks containing spent nuclear fuel (SNF) that are stored for hundreds of thousands of years in high-level wastes (HLW) and SNF repositories (e.g. Yucca Mountain Project). In this case DU oxides serve as an additional engi- neered chemical barrier. This work is a part of the joint Russian –American Program on Beneficial Use of Depleted Uranium. This paper describes the UO2 transformations that take place in contact with various aqueous media (deionized water (DW), J-13 solution that simulates Yucca Mountain ground water, etc.) and sorption of long-lived radionuclides (237Np and 99Тс) from these media by depleted uranium dioxide. Samples of depleted uranium dioxide used in this work originated from the treatment of UF6 in a reducing media to form UO2 (DUO2-1 at 600°C, DUO2-2 at 700°C, and DUO2-3 at 800°C).