Irreversible Oxidation of Protein Cysteine Residues Michael James Hamann Iowa State University
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Effect of Peptide Histidine Isoleucine on Water and Electrolyte Transport in the Human Jejunum
Gut: first published as 10.1136/gut.25.6.624 on 1 June 1984. Downloaded from Gut, 1984, 25, 624-628 Alimentary tract and pancreas Effect of peptide histidine isoleucine on water and electrolyte transport in the human jejunum K J MORIARTY, J E HEGARTY, K TATEMOTO, V MUTT, N D CHRISTOFIDES, S R BLOOM, AND J R WOOD From the Department of Gastroenterology, St Bartholomew's Hospital, London, The Liver Unit, King's College Hospital, London, Department ofMedicine, Hammersmith Hospital, London, and Department of Biochemistry, Karolinska Institute, Stockholm, Sweden SUMMARY Peptide histidine isoleucine, a 27 amino acid peptide with close amino acid sequence homology to vasoactive intestinal peptide and secretin, is distributed throughout the mammalian intestinal tract, where it has been localised to intramural neurones. An intestinal perfusion technique has been used to study the effect of intravenous peptide histidine isoleucine (44.5 pmol/kg/min) on water and electrolyte transport from a plasma like electrolyte solution in human jejunum in vivo. Peptide histidine isoleucine infusion produced peak plasma peptide histidine isoleucine concentrations in the range 2000-3000 pmolIl, flushing, tachycardia and a reduction in diastolic blood pressure. Peptide histidine isoleucine caused a significant inhibition of net absorption of water, sodium, potassium and bicarbonate and induced a net secretion of chloride, these changes being completely reversed during the post-peptide histidine isoleucine period. These findings suggest that endogenous peptide histidine isoleucine may participate in the neurohumoral regulation of water and electrolyte transport in the human jejunum. http://gut.bmj.com/ Peptide histidine isoleucine, isolated originally from INTESTINAL PERFUSION mammalian small intestine, is a 27-amino acid After an eight hour fast, each subject swallowed a peptide having close amino acid sequence homology double lumen intestinal perfusion tube, incorpo- to vasoactive intestinal peptide and secretin. -

Isoleucine, an Essential Amino Acid, Prevents Liver Metastases of Colon Cancer by Antiangiogenesis Kazumoto Murata1 and Masami Moriyama2
Research Article Isoleucine, an Essential Amino Acid, Prevents Liver Metastases of Colon Cancer by Antiangiogenesis Kazumoto Murata1 and Masami Moriyama2 1The First Department of Internal Medicine, Mie University School of Medicine, Tsu, Mie, Japan and 2Microbiology and Immunology, Keio University School of Medicine, Tokyo, Japan Abstract infection in the airways of cystic fibrosis (8), and susceptibility to salmonella infection in mouse intestinal tracts (9). In addition to In spite of recent advances in the treatment of colon cancer, h multiple liver metastases of colon cancer are still difficult to their direct antimicrobial activities, -defensins are strong treat. Some chemotherapeutic regimens have been reported to chemotactic factors for memory T cells and dendritic cells, be efficient, but there is a high risk of side effects associated suggesting that they also play an important role in acquired immunity (10–12). h-defensins are also inducible by inflammatory with these. Here, we show that isoleucine, an essential amino a h acid, prevents liver metastases in a mouse colon cancer cytokines, such as tumor necrosis factor- and interleukin-1 (13, 14). Recently, Fehlbaum et al. (15) reported that isoleucine metastatic model. Because isoleucine is a strong inducer of h B-defensin, we first hypothesized that it prevented liver and its analogues are highly specific inducers of -defensins. We thus originally hypothesized that isoleucine may contribute to metastases via the accumulation of dendritic cells or memory B tumor immunity through both innate and acquired immunity by T cells through up-regulation of -defensin. However, neither h B-defensin nor immunologic responses were induced by induction of -defensins. -
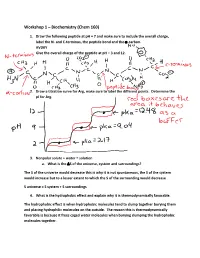
Workshop 1 – Biochemistry (Chem 160)
Workshop 1 – Biochemistry (Chem 160) 1. Draw the following peptide at pH = 7 and make sure to include the overall charge, label the N- and C-terminus, the peptide bond and the -carbon. AVDKY Give the overall charge of the peptide at pH = 3 and 12. 2. Draw a titration curve for Arg, make sure to label the different points. Determine the pI for Arg. 3. Nonpolar solute + water = solution a. What is the S of the universe, system and surroundings? The S of the universe would decrease this is why it is not spontaneous, the S of the system would increase but to a lesser extent to which the S of the surrounding would decrease S universe = S system + S surroundings 4. What is the hydrophobic effect and explain why it is thermodynamically favorable. The hydrophobic effect is when hydrophobic molecules tend to clump together burying them and placing hydrophilic molecules on the outside. The reason this is thermodynamically favorable is because it frees caged water molecules when burying clumping the hydrophobic molecules together. 5. Urea dissolves very readily in water, but the solution becomes very cold as the urea dissolves. How is this possible? Urea dissolves in water because when dissolving there is a net increase in entropy of the universe. The heat exchange, getting colder only reflects the enthalpy (H) component of the total energy change. The entropy change is high enough to offset the enthalpy component and to add up to an overall -G 6. A mutation that changes an alanine residue in the interior of a protein to valine is found to lead to a loss of activity. -

The Optimum Isoleucine:Lysine Ratio in Starter Diets to Maximize Growth Performance of the Early-Weaned Pig
Kansas Agricultural Experiment Station Research Reports Volume 0 Issue 10 Swine Day (1968-2014) Article 837 2000 The optimum isoleucine:lysine ratio in starter diets to maximize growth performance of the early-weaned pig B W. James Robert D. Goodband Michael D. Tokach See next page for additional authors Follow this and additional works at: https://newprairiepress.org/kaesrr Part of the Other Animal Sciences Commons Recommended Citation James, B W.; Goodband, Robert D.; Tokach, Michael D.; Nelssen, Jim L.; and DeRouchey, Joel M. (2000) "The optimum isoleucine:lysine ratio in starter diets to maximize growth performance of the early-weaned pig," Kansas Agricultural Experiment Station Research Reports: Vol. 0: Iss. 10. https://doi.org/10.4148/ 2378-5977.6677 This report is brought to you for free and open access by New Prairie Press. It has been accepted for inclusion in Kansas Agricultural Experiment Station Research Reports by an authorized administrator of New Prairie Press. Copyright 2000 Kansas State University Agricultural Experiment Station and Cooperative Extension Service. Contents of this publication may be freely reproduced for educational purposes. All other rights reserved. Brand names appearing in this publication are for product identification purposes only. No endorsement is intended, nor is criticism implied of similar products not mentioned. K-State Research and Extension is an equal opportunity provider and employer. The optimum isoleucine:lysine ratio in starter diets to maximize growth performance of the early-weaned pig Abstract A total of 360 weanling pigs (initially 12.3 lb BW and approximately 18 d of age) was used in a 14-d growth assay to determine the optimal isoleucine:lysine ratio to maximize growth performance. -

Mechanistic Insights on the Reduction of Glutathione Disulfide by Protein Disulfide Isomerase
Mechanistic insights on the reduction of glutathione disulfide by protein disulfide isomerase Rui P. P. Nevesa, Pedro Alexandrino Fernandesa, and Maria João Ramosa,1 aUnidade de Ciências Biomoleculares Aplicadas, Rede de Química e Tecnologia, Departamento de Química e Bioquímica, Faculdade de Ciências, Universidade do Porto, 4169-007 Porto, Portugal Edited by Donald G. Truhlar, University of Minnesota, Minneapolis, MN, and approved May 9, 2017 (received for review November 22, 2016) We explore the enzymatic mechanism of the reduction of glutathione of enzymes, which are responsible for the reduction and isomer- disulfide (GSSG) by the reduced a domain of human protein disulfide ization of disulfide bonds, through thiol-disulfide exchange. isomerase (hPDI) with atomistic resolution. We use classical molecular dynamics and hybrid quantum mechanics/molecular mechanics cal- Structure and Function of PDI culations at the mPW1N/6–311+G(2d,2p):FF99SB//mPW1N/6–31G(d): Human protein disulfide isomerase (hPDI) is a U-shaped enzyme FF99SB level. The reaction proceeds in two stages: (i) a thiol-disulfide with 508 residues. Its tertiary structure is composed of four exchange through nucleophilic attack of the Cys53-thiolate to the thioredoxin-like domains (a, b, b′,anda′) and a fifth tail-shaped c GSSG-disulfide followed by the deprotonation of Cys56-thiol by domain (Fig. 1) (14, 15). The maximum activity of hPDI is observed Glu47-carboxylate and (ii) a second thiol-disulfide exchange between when all domains of PDI contribute synergistically to its function (16). the Cys56-thiolate and the mixed disulfide intermediate formed in Similar to thioredoxin, the a and a′ domains have a catalytic the first step. -

Degradation of Glutathione in Plant Cells
Degradation of Glutathione in Plant Cells: Evidence against the Participation of a y-Glutamyltranspeptidase Reinhard Steinkamp and Heinz Rennenberg Botanisches Institut der Universität zu Köln, Gyrhofstr. 15, D-5000 Köln 41, Bundesrepublik Deutschland Z. Naturforsch. 40c, 29 — 33 (1985); received August 31/October 4, 1984 Tobacco, Glutathione Catabolism, y-Glutamylcysteine, y-Glutamyltranspeptidase, y-Glutamyl- cyclotransferase When y-glutamyltranspeptidase activity in tobacco cells was measured using the artificial substrate y-glutamyl-/?-nitroanilide, liberation of p-nitroaniline was not reduced, but stimulated by addition of glutathione. Therefore, glutathione was not acting as a donator, but as an acceptor of y-glutamyl moieties in the assay mixture, suggesting that y-glutamyltranspeptidase is not participating in degradation of glutathione. Feeding experiments with [^S-cysJglutathione sup ported this conclusion. When tobacco cells were supplied with this peptide as sole sulfur source, glutathione and y-glutamylcysteine were the only labelled compounds found inside the cells. The low rate of uptake of glutathione apparently prevented the accumulation of measurable amounts of radioactivity in the cysteine pool. A y-glutamylcyclotransferase, responsible for the conversion of y-glutamylcysteine to 5-oxo-proline and cysteine was found in ammonium sulfate precipitates of tobacco cell homogenates. The enzyme showed high activities with y-glutamylmethionine and y-glutamylcysteine, but not with other y-glutamyldipeptides or glutathione. From these and previously published experiments [(Rennenberg et al., Z. Naturforsch. 3 5 c, 70 8 -7 1 1 (1980)], it is concluded that glutathione is degraded in tobacco cells via the following pathway: y-glu-cys- gly —> y-glu-cys ->• 5-oxo-proline -* glu. Introduction the cysteine conjugate by the action of a y-gluta myltranspeptidase (Fig. -

Sulfhydryl Reduction of Methylene Blue with Reference to Alterations in Malignant Neoplastic Disease
Sulfhydryl Reduction of Methylene Blue With Reference to Alterations in Malignant Neoplastic Disease Maurice M. Black, M. D. (From the Department of Biochemistry, New York Medical College, New York 29, N. t;., and the Brooklyn Cancer Institute, Brooklyn 9, N. Y.) (Received for publication May 8, 1947) A significant decrease in methylene blue re- reactivity is less than half that of the cysteine. It is ducing power of plasma from patients with malig- noteworthy also that the resultant leuco mixture nant neoplastic disease was previously reported did not revert back to colored methylene blue on (1). At that time it was suggested that change in a cooling, as was the case with methylene blue re- reducing group of the albumin molecule was a duction by plasma. likely source of this alteration. Similar conclusions Similar relationships were investigated between were reported also by Savignac and associates (7) cysteine and different concentrations of methylene as the result of analogous studies. blue. As seen in Fig. 2, similar curves are obtained, In an attempt to evaluate the effect of the sulf- but the position of the curve on the graph varies hydryl group on the reduction of methylene blue, a with the concentration of the methylene blue used. study was undertaken with various compounds of It should be noted that there is no appreciable known -SH and S-S structures. In addition, an difference in the reducing time of methylene blue attempt was made to establish a standard method on varying the concentrations between 0.10 per of calibration of various lots of methylene blue, so cent and 0.2 per cent, although 0.08 per cent shows that more uniform results would be possible in the a decided difference. -

Effect of Leucine on Intestinal Absorption of Tryptophan in Rats
Downloaded from https://doi.org/10.1079/BJN19850155 British Journal of Nutrition (1985), 54, 695-703 695 https://www.cambridge.org/core Effect of leucine on intestinal absorption of tryptophan in rats BY CHISAE UMEZAWA, YUKO MAEDA, KANJI HABA, MARIKO SHIN AND KEIJI SANO School of Pharmacy, Kobe-Gakuin University, Nishi-ku, Kobe 673, Japan (Received I7 May 1985 - Accepted 24 June 1985) . IP address: 1. To elucidate the causal relation between leucine and the lowering of hepatic NAD content of rats fed on a leucine-excessive diet (Yamada et aZ. 1979), the effect of leucine on intestinal absorption of tryptophan was 170.106.35.93 investigated. 2. Co-administration of [3H]tryptophan and leucine, with leucine at ten times the level of tryptophan, delayed absorption of L-[side chain 2,3-3H]tryptophan from the digestive tract and incorporation of [3H]tryptophan into portal blood, the liver and a protein fraction of the liver. After 120 min, more than 95% of tryptophan was absorbed whether [3H]tryptophan was administered with or without leucine. , on 3. Co-administration of a mixture of ten essential amino acids, in proportions simulating casein, with 02 Oct 2021 at 04:49:27 [3H]tryptophan markedly delayed absorption of tryptophan from the digestive tract. The addition of supplementary leucine to the amino acid mixture, however, caused no further delay. 4. In rats prefed a leucine-excessive diet for 1 week [3H]tryptophan was absorbed at the same rate as in rats fed on a control diet. 5. The results indicate that competition between tryptophan and leucine for intestinal absorption did not cause lowering of hepatic NAD. -

A Review of Dietary (Phyto)Nutrients for Glutathione Support
nutrients Review A Review of Dietary (Phyto)Nutrients for Glutathione Support Deanna M. Minich 1,* and Benjamin I. Brown 2 1 Human Nutrition and Functional Medicine Graduate Program, University of Western States, 2900 NE 132nd Ave, Portland, OR 97230, USA 2 BCNH College of Nutrition and Health, 116–118 Finchley Road, London NW3 5HT, UK * Correspondence: [email protected] Received: 8 July 2019; Accepted: 23 August 2019; Published: 3 September 2019 Abstract: Glutathione is a tripeptide that plays a pivotal role in critical physiological processes resulting in effects relevant to diverse disease pathophysiology such as maintenance of redox balance, reduction of oxidative stress, enhancement of metabolic detoxification, and regulation of immune system function. The diverse roles of glutathione in physiology are relevant to a considerable body of evidence suggesting that glutathione status may be an important biomarker and treatment target in various chronic, age-related diseases. Yet, proper personalized balance in the individual is key as well as a better understanding of antioxidants and redox balance. Optimizing glutathione levels has been proposed as a strategy for health promotion and disease prevention, although clear, causal relationships between glutathione status and disease risk or treatment remain to be clarified. Nonetheless, human clinical research suggests that nutritional interventions, including amino acids, vitamins, minerals, phytochemicals, and foods can have important effects on circulating glutathione which may translate to clinical benefit. Importantly, genetic variation is a modifier of glutathione status and influences response to nutritional factors that impact glutathione levels. This narrative review explores clinical evidence for nutritional strategies that could be used to improve glutathione status. -

Electrochemical Studies of Dl-Leucine, L-Proline and L
ELECTROCHEMICAL STUDIES OF DL -LEUCINE, 60 L-PROLINE AND L-TRYPTOPHAN AND THEIR INTERACTION WITH COPPER AND IRON 30 c b A) M. A. Jabbar, R. J. Mannan, S. Salauddin and B. µ a Rashid 0 Department of Chemistry, University of Dhaka, ( Current Dhaka-1000, Bangladesh -30 Introduction -60 In vitro study of the charge transfer reactions coupled -800 -400 0 400 800 with chemical reactions can give important indication of Potential vs. Ag/AgCl (mV) about actual biological processes occurring in human Fig.1. Comparison of the cyclic voltammogram of system. Understanding of such charge-transfer 5.0mM (a) DL -Leucine, (b) Cu-DL -Leucine ion and mechanism will help to determine the effectiveness of (c) [Fe-DL -Leucine] in 0.1M KCl solution at a Pt- nutrition, metabolism and treatment of various biological button electrode. Scan rates 50 mV/s. disorders. In the previous research, the redox behaviour of 40 various amino acids and biochemically important compounds and their charge transfer reaction and their b interaction of metal ions were studied [1,2]. In the present 20 ) a c research, the redox behavior and the charge transfer µΑ kinetics of DL -Leucine, L-Proline and L-Tryptophan in 0 the presence and absence of copper and iron will be investigated. Current ( -20 Experimental A computerized electrochemistry system developed by -40 -800 -400 0 400 800 Advanced Analytics, Virginia, USA, (Model-2040) Potential vs. Ag/AgCl (mV) consisting of three electrodes micro-cell with a saturated Ag/AgCl reference, a Pt-wire auxiliary and a pretreated Fig.2 . Comparison of the cyclic voltammogram of Pt-button working electrode is employed to investigate 5.0mM (a) L-Proline, (b) Cu-L-Proline and (c) Fe-L- Proline in 0.1M KCl solution at a Pt-button different amino acids and metal-amino acid systems. -

Muscle Wasting and Aging: Experimental Models, Fatty Infiltrations, and Prevention Thomas Brioche, Allan Pagano, Guillaume Py, Angèle Chopard
Muscle wasting and aging: Experimental models, fatty infiltrations, and prevention Thomas Brioche, Allan Pagano, Guillaume Py, Angèle Chopard To cite this version: Thomas Brioche, Allan Pagano, Guillaume Py, Angèle Chopard. Muscle wasting and aging: Experi- mental models, fatty infiltrations, and prevention. Molecular Aspects of Medicine, Elsevier, 2016,32 p. 10.1016/j.mam.2016.04.006. hal-01837630 HAL Id: hal-01837630 https://hal.archives-ouvertes.fr/hal-01837630 Submitted on 28 May 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Distributed under a Creative Commons Attribution - ShareAlike| 4.0 International License Accepted Manuscript Title: Muscle wasting and aging: experimental models, fatty infiltrations, and prevention Author: Thomas Brioche, Allan F. Pagano, Guillaume Py, Angèle Chopard PII: S0098-2997(15)30021-2 DOI: http://dx.doi.org/doi: 10.1016/j.mam.2016.04.006 Reference: JMAM 642 To appear in: Molecular Aspects of Medicine Received date: 19-12-2015 Revised date: 13-4-2016 Accepted date: 13-4-2016 Please cite this article as: Thomas Brioche, Allan F. Pagano, Guillaume Py, Angèle Chopard, Muscle wasting and aging: experimental models, fatty infiltrations, and prevention, Molecular Aspects of Medicine (2016), http://dx.doi.org/doi: 10.1016/j.mam.2016.04.006. -

Amino Acid Chemistry
Handout 4 Amino Acid and Protein Chemistry ANSC 619 PHYSIOLOGICAL CHEMISTRY OF LIVESTOCK SPECIES Amino Acid Chemistry I. Chemistry of amino acids A. General amino acid structure + HN3- 1. All amino acids are carboxylic acids, i.e., they have a –COOH group at the #1 carbon. 2. All amino acids contain an amino group at the #2 carbon (may amino acids have a second amino group). 3. All amino acids are zwitterions – they contain both positive and negative charges at physiological pH. II. Essential and nonessential amino acids A. Nonessential amino acids: can make the carbon skeleton 1. From glycolysis. 2. From the TCA cycle. B. Nonessential if it can be made from an essential amino acid. 1. Amino acid "sparing". 2. May still be essential under some conditions. C. Essential amino acids 1. Branched chain amino acids (isoleucine, leucine and valine) 2. Lysine 3. Methionine 4. Phenyalanine 5. Threonine 6. Tryptophan 1 Handout 4 Amino Acid and Protein Chemistry D. Essential during rapid growth or for optimal health 1. Arginine 2. Histidine E. Nonessential amino acids 1. Alanine (from pyruvate) 2. Aspartate, asparagine (from oxaloacetate) 3. Cysteine (from serine and methionine) 4. Glutamate, glutamine (from α-ketoglutarate) 5. Glycine (from serine) 6. Proline (from glutamate) 7. Serine (from 3-phosphoglycerate) 8. Tyrosine (from phenylalanine) E. Nonessential and not required for protein synthesis 1. Hydroxyproline (made postranslationally from proline) 2. Hydroxylysine (made postranslationally from lysine) III. Acidic, basic, polar, and hydrophobic amino acids A. Acidic amino acids: amino acids that can donate a hydrogen ion (proton) and thereby decrease pH in an aqueous solution 1.