Supplemental Table 1. Exome Variant Filtering Strategy a Based on UCSC
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Seq2pathway Vignette
seq2pathway Vignette Bin Wang, Xinan Holly Yang, Arjun Kinstlick May 19, 2021 Contents 1 Abstract 1 2 Package Installation 2 3 runseq2pathway 2 4 Two main functions 3 4.1 seq2gene . .3 4.1.1 seq2gene flowchart . .3 4.1.2 runseq2gene inputs/parameters . .5 4.1.3 runseq2gene outputs . .8 4.2 gene2pathway . 10 4.2.1 gene2pathway flowchart . 11 4.2.2 gene2pathway test inputs/parameters . 11 4.2.3 gene2pathway test outputs . 12 5 Examples 13 5.1 ChIP-seq data analysis . 13 5.1.1 Map ChIP-seq enriched peaks to genes using runseq2gene .................... 13 5.1.2 Discover enriched GO terms using gene2pathway_test with gene scores . 15 5.1.3 Discover enriched GO terms using Fisher's Exact test without gene scores . 17 5.1.4 Add description for genes . 20 5.2 RNA-seq data analysis . 20 6 R environment session 23 1 Abstract Seq2pathway is a novel computational tool to analyze functional gene-sets (including signaling pathways) using variable next-generation sequencing data[1]. Integral to this tool are the \seq2gene" and \gene2pathway" components in series that infer a quantitative pathway-level profile for each sample. The seq2gene function assigns phenotype-associated significance of genomic regions to gene-level scores, where the significance could be p-values of SNPs or point mutations, protein-binding affinity, or transcriptional expression level. The seq2gene function has the feasibility to assign non-exon regions to a range of neighboring genes besides the nearest one, thus facilitating the study of functional non-coding elements[2]. Then the gene2pathway summarizes gene-level measurements to pathway-level scores, comparing the quantity of significance for gene members within a pathway with those outside a pathway. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -
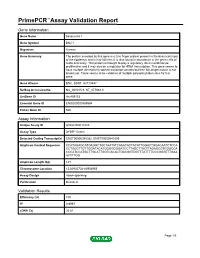
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name basonuclin 1 Gene Symbol BNC1 Organism Human Gene Summary The protein encoded by this gene is a zinc finger protein present in the basal cell layer of the epidermis and in hair follicles. It is also found in abundance in the germ cells of testis and ovary. This protein is thought to play a regulatory role in keratinocyte proliferation and it may also be a regulator for rRNA transcription. This gene seems to have multiple alternatively spliced transcript variants but their full-length nature is not known yet. There seems to be evidence of multiple polyadenylation sites for this gene. Gene Aliases BNC, BSN1, HsT19447 RefSeq Accession No. NC_000015.9, NT_077661.3 UniGene ID Hs.459153 Ensembl Gene ID ENSG00000169594 Entrez Gene ID 646 Assay Information Unique Assay ID qHsaCID0017223 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000345382, ENST00000541809 Amplicon Context Sequence CCATAGAGCATGAGGCTGCTAATATCAAACACTACATTGGACTGGACAATCTCCA CCTGGCTTGTTGGATACATGGGGGGGATCCTTAGCTTACTTAGAGCGTGGGCCA CCCATCCATGCTTGCATTGGTCACACTGACGGTGGTTTATTTTCCCGGGTTTGAA ACTTTGG Amplicon Length (bp) 141 Chromosome Location 15:83935724-83936955 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 100 R2 0.9997 cDNA Cq 26.81 Page 1/5 PrimePCR™Assay Validation Report cDNA Tm (Celsius) 84.5 gDNA Cq 39.42 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 2/5 PrimePCR™Assay Validation Report BNC1, Human Amplification -

Proteomics Provides Insights Into the Inhibition of Chinese Hamster V79
www.nature.com/scientificreports OPEN Proteomics provides insights into the inhibition of Chinese hamster V79 cell proliferation in the deep underground environment Jifeng Liu1,2, Tengfei Ma1,2, Mingzhong Gao3, Yilin Liu4, Jun Liu1, Shichao Wang2, Yike Xie2, Ling Wang2, Juan Cheng2, Shixi Liu1*, Jian Zou1,2*, Jiang Wu2, Weimin Li2 & Heping Xie2,3,5 As resources in the shallow depths of the earth exhausted, people will spend extended periods of time in the deep underground space. However, little is known about the deep underground environment afecting the health of organisms. Hence, we established both deep underground laboratory (DUGL) and above ground laboratory (AGL) to investigate the efect of environmental factors on organisms. Six environmental parameters were monitored in the DUGL and AGL. Growth curves were recorded and tandem mass tag (TMT) proteomics analysis were performed to explore the proliferative ability and diferentially abundant proteins (DAPs) in V79 cells (a cell line widely used in biological study in DUGLs) cultured in the DUGL and AGL. Parallel Reaction Monitoring was conducted to verify the TMT results. γ ray dose rate showed the most detectable diference between the two laboratories, whereby γ ray dose rate was signifcantly lower in the DUGL compared to the AGL. V79 cell proliferation was slower in the DUGL. Quantitative proteomics detected 980 DAPs (absolute fold change ≥ 1.2, p < 0.05) between V79 cells cultured in the DUGL and AGL. Of these, 576 proteins were up-regulated and 404 proteins were down-regulated in V79 cells cultured in the DUGL. KEGG pathway analysis revealed that seven pathways (e.g. -

FMRP Links Optimal Codons to Mrna Stability in Neurons
FMRP links optimal codons to mRNA stability in neurons Huan Shua,1, Elisa Donnardb, Botao Liua, Suna Junga, Ruijia Wanga, and Joel D. Richtera aProgram in Molecular Medicine, University of Massachusetts Medical School, Worcester, MA 01605; and bBioinformatics and Integrative Biology, University of Massachusetts Medical School, Worcester, MA 01605 Edited by Lynne E. Maquat, University of Rochester School of Medicine and Dentistry, Rochester, NY, and approved October 12, 2020 (received for review May 8, 2020) Fragile X syndrome (FXS) is caused by inactivation of the FMR1 Here, we have used ribosome profiling and RNA sequencing gene and loss of encoded FMRP, an RNA binding protein that re- (RNA-seq) to investigate translational dysregulation in the presses translation of some of its target transcripts. Here we use FMRP KO cortex and found that FMRP coordinates the link ribosome profiling and RNA sequencing to investigate the dysre- between RNA destruction and codon usage bias (codon opti- gulation of translation in the mouse brain cortex. We find that mality). We find that the apparent dysregulation of translational most changes in ribosome occupancy on hundreds of mRNAs are activity (i.e., ribosome occupancy) in FMRP KO cortex can be largely driven by dysregulation in transcript abundance. Many accounted for by commensurate changes in steady-state RNA down-regulated mRNAs, which are mostly responsible for neuro- levels. Down-regulated mRNAs in FMRP KO cortex are nal and synaptic functions, are highly enriched for FMRP binding enriched for those that encode factors involved in neuronal and targets. RNA metabolic labeling demonstrates that, in FMRP- synaptic functions and are highly enriched for FMRP binding deficient cortical neurons, mRNA down-regulation is caused by targets. -

Transcriptome Sequencing and Genome-Wide Association Analyses Reveal Lysosomal Function and Actin Cytoskeleton Remodeling in Schizophrenia and Bipolar Disorder
Molecular Psychiatry (2015) 20, 563–572 © 2015 Macmillan Publishers Limited All rights reserved 1359-4184/15 www.nature.com/mp ORIGINAL ARTICLE Transcriptome sequencing and genome-wide association analyses reveal lysosomal function and actin cytoskeleton remodeling in schizophrenia and bipolar disorder Z Zhao1,6,JXu2,6, J Chen3,6, S Kim4, M Reimers3, S-A Bacanu3,HYu1, C Liu5, J Sun1, Q Wang1, P Jia1,FXu2, Y Zhang2, KS Kendler3, Z Peng2 and X Chen3 Schizophrenia (SCZ) and bipolar disorder (BPD) are severe mental disorders with high heritability. Clinicians have long noticed the similarities of clinic symptoms between these disorders. In recent years, accumulating evidence indicates some shared genetic liabilities. However, what is shared remains elusive. In this study, we conducted whole transcriptome analysis of post-mortem brain tissues (cingulate cortex) from SCZ, BPD and control subjects, and identified differentially expressed genes in these disorders. We found 105 and 153 genes differentially expressed in SCZ and BPD, respectively. By comparing the t-test scores, we found that many of the genes differentially expressed in SCZ and BPD are concordant in their expression level (q ⩽ 0.01, 53 genes; q ⩽ 0.05, 213 genes; q ⩽ 0.1, 885 genes). Using genome-wide association data from the Psychiatric Genomics Consortium, we found that these differentially and concordantly expressed genes were enriched in association signals for both SCZ (Po10 − 7) and BPD (P = 0.029). To our knowledge, this is the first time that a substantially large number of genes show concordant expression and association for both SCZ and BPD. Pathway analyses of these genes indicated that they are involved in the lysosome, Fc gamma receptor-mediated phagocytosis, regulation of actin cytoskeleton pathways, along with several cancer pathways. -

Supplemental Information
Supplemental information Dissection of the genomic structure of the miR-183/96/182 gene. Previously, we showed that the miR-183/96/182 cluster is an intergenic miRNA cluster, located in a ~60-kb interval between the genes encoding nuclear respiratory factor-1 (Nrf1) and ubiquitin-conjugating enzyme E2H (Ube2h) on mouse chr6qA3.3 (1). To start to uncover the genomic structure of the miR- 183/96/182 gene, we first studied genomic features around miR-183/96/182 in the UCSC genome browser (http://genome.UCSC.edu/), and identified two CpG islands 3.4-6.5 kb 5’ of pre-miR-183, the most 5’ miRNA of the cluster (Fig. 1A; Fig. S1 and Seq. S1). A cDNA clone, AK044220, located at 3.2-4.6 kb 5’ to pre-miR-183, encompasses the second CpG island (Fig. 1A; Fig. S1). We hypothesized that this cDNA clone was derived from 5’ exon(s) of the primary transcript of the miR-183/96/182 gene, as CpG islands are often associated with promoters (2). Supporting this hypothesis, multiple expressed sequences detected by gene-trap clones, including clone D016D06 (3, 4), were co-localized with the cDNA clone AK044220 (Fig. 1A; Fig. S1). Clone D016D06, deposited by the German GeneTrap Consortium (GGTC) (http://tikus.gsf.de) (3, 4), was derived from insertion of a retroviral construct, rFlpROSAβgeo in 129S2 ES cells (Fig. 1A and C). The rFlpROSAβgeo construct carries a promoterless reporter gene, the β−geo cassette - an in-frame fusion of the β-galactosidase and neomycin resistance (Neor) gene (5), with a splicing acceptor (SA) immediately upstream, and a polyA signal downstream of the β−geo cassette (Fig. -

The DNA Methylation Landscape of Glioblastoma Disease Progression Shows Extensive Heterogeneity in Time and Space
bioRxiv preprint doi: https://doi.org/10.1101/173864; this version posted August 9, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. The DNA methylation landscape of glioblastoma disease progression shows extensive heterogeneity in time and space Johanna Klughammer1*, Barbara Kiesel2,3*, Thomas Roetzer3,4, Nikolaus Fortelny1, Amelie Kuchler1, Nathan C. Sheffield5, Paul Datlinger1, Nadine Peter3,4, Karl-Heinz Nenning6, Julia Furtner3,7, Martha Nowosielski8,9, Marco Augustin10, Mario Mischkulnig2,3, Thomas Ströbel3,4, Patrizia Moser11, Christian F. Freyschlag12, Jo- hannes Kerschbaumer12, Claudius Thomé12, Astrid E. Grams13, Günther Stockhammer8, Melitta Kitzwoegerer14, Stefan Oberndorfer15, Franz Marhold16, Serge Weis17, Johannes Trenkler18, Johanna Buchroithner19, Josef Pichler20, Johannes Haybaeck21,22, Stefanie Krassnig21, Kariem Madhy Ali23, Gord von Campe23, Franz Payer24, Camillo Sherif25, Julius Preiser26, Thomas Hauser27, Peter A. Winkler27, Waltraud Kleindienst28, Franz Würtz29, Tanisa Brandner-Kokalj29, Martin Stultschnig30, Stefan Schweiger31, Karin Dieckmann3,32, Matthias Preusser3,33, Georg Langs6, Bernhard Baumann10, Engelbert Knosp2,3, Georg Widhalm2,3, Christine Marosi3,33, Johannes A. Hainfellner3,4, Adelheid Woehrer3,4#§, Christoph Bock1,34,35# 1 CeMM Research Center for Molecular Medicine of the Austrian Academy of Sciences, Vienna, Austria. 2 Department of Neurosurgery, Medical University of Vienna, Vienna, Austria. 3 Comprehensive Cancer Center, Central Nervous System Tumor Unit, Medical University of Vienna, Austria. 4 Institute of Neurology, Medical University of Vienna, Vienna, Austria. 5 Center for Public Health Genomics, University of Virginia, Charlottesville VA, USA. 6 Department of Biomedical Imaging and Image-guided Therapy, Computational Imaging Research Lab, Medical University of Vi- enna, Vienna, Austria. -

NICU Gene List Generator.Xlsx
Neonatal Crisis Sequencing Panel Gene List Genes: A2ML1 - B3GLCT A2ML1 ADAMTS9 ALG1 ARHGEF15 AAAS ADAMTSL2 ALG11 ARHGEF9 AARS1 ADAR ALG12 ARID1A AARS2 ADARB1 ALG13 ARID1B ABAT ADCY6 ALG14 ARID2 ABCA12 ADD3 ALG2 ARL13B ABCA3 ADGRG1 ALG3 ARL6 ABCA4 ADGRV1 ALG6 ARMC9 ABCB11 ADK ALG8 ARPC1B ABCB4 ADNP ALG9 ARSA ABCC6 ADPRS ALK ARSL ABCC8 ADSL ALMS1 ARX ABCC9 AEBP1 ALOX12B ASAH1 ABCD1 AFF3 ALOXE3 ASCC1 ABCD3 AFF4 ALPK3 ASH1L ABCD4 AFG3L2 ALPL ASL ABHD5 AGA ALS2 ASNS ACAD8 AGK ALX3 ASPA ACAD9 AGL ALX4 ASPM ACADM AGPS AMELX ASS1 ACADS AGRN AMER1 ASXL1 ACADSB AGT AMH ASXL3 ACADVL AGTPBP1 AMHR2 ATAD1 ACAN AGTR1 AMN ATL1 ACAT1 AGXT AMPD2 ATM ACE AHCY AMT ATP1A1 ACO2 AHDC1 ANK1 ATP1A2 ACOX1 AHI1 ANK2 ATP1A3 ACP5 AIFM1 ANKH ATP2A1 ACSF3 AIMP1 ANKLE2 ATP5F1A ACTA1 AIMP2 ANKRD11 ATP5F1D ACTA2 AIRE ANKRD26 ATP5F1E ACTB AKAP9 ANTXR2 ATP6V0A2 ACTC1 AKR1D1 AP1S2 ATP6V1B1 ACTG1 AKT2 AP2S1 ATP7A ACTG2 AKT3 AP3B1 ATP8A2 ACTL6B ALAS2 AP3B2 ATP8B1 ACTN1 ALB AP4B1 ATPAF2 ACTN2 ALDH18A1 AP4M1 ATR ACTN4 ALDH1A3 AP4S1 ATRX ACVR1 ALDH3A2 APC AUH ACVRL1 ALDH4A1 APTX AVPR2 ACY1 ALDH5A1 AR B3GALNT2 ADA ALDH6A1 ARFGEF2 B3GALT6 ADAMTS13 ALDH7A1 ARG1 B3GAT3 ADAMTS2 ALDOB ARHGAP31 B3GLCT Updated: 03/15/2021; v.3.6 1 Neonatal Crisis Sequencing Panel Gene List Genes: B4GALT1 - COL11A2 B4GALT1 C1QBP CD3G CHKB B4GALT7 C3 CD40LG CHMP1A B4GAT1 CA2 CD59 CHRNA1 B9D1 CA5A CD70 CHRNB1 B9D2 CACNA1A CD96 CHRND BAAT CACNA1C CDAN1 CHRNE BBIP1 CACNA1D CDC42 CHRNG BBS1 CACNA1E CDH1 CHST14 BBS10 CACNA1F CDH2 CHST3 BBS12 CACNA1G CDK10 CHUK BBS2 CACNA2D2 CDK13 CILK1 BBS4 CACNB2 CDK5RAP2 -

Identification of Novel DNA-Methylated Genes
Prostate Cancer and Prostatic Disease (2013) 16, 292–300 & 2013 Macmillan Publishers Limited All rights reserved 1365-7852/13 www.nature.com/pcan ORIGINAL ARTICLE Identification of novel DNA-methylated genes that correlate with human prostate cancer and high-grade prostatic intraepithelial neoplasia JM Devaney1, S Wang2,3, S Funda1, J Long2, DJ Taghipour2, R Tbaishat3, P Furbert-Harris2,4, M Ittmann5 and B Kwabi-Addo2,3 BACKGROUND: Prostate cancer (PCa) harbors a myriad of genomic and epigenetic defects. Cytosine methylation of CpG-rich promoter DNA is an important mechanism of epigenetic gene inactivation in PCa. There is considerable amount of data to suggest that DNA methylation-based biomarkers may be useful for the early detection and diagnosis of PCa. In addition, candidate gene- based studies have shown an association between specific gene methylation and alterations and clinicopathologic indicators of poor prognosis in PCa. METHODS: To more comprehensively identify DNA methylation alterations in PCa initiation and progression, we examined the methylation status of 485 577 CpG sites from regions with a broad spectrum of CpG densities, interrogating both gene-associated and non-associated regions using the recently developed Illumina 450K methylation platform. RESULTS: In all, we selected 33 promoter-associated novel CpG sites that were differentially methylated in high-grade prostatic intraepithelial neoplasia and PCa in comparison with benign prostate tissue samples (false discovery rate-adjusted P-value o0.05; b-value X0.2; fold change 41.5). Of the 33 genes, hierarchical clustering analysis demonstrated BNC1, FZD1, RPL39L, SYN2, LMX1B, CXXC5, ZNF783 and CYB5R2 as top candidate novel genes that are frequently methylated and whose methylation was associated with inactivation of gene expression in PCa cell lines. -

Noelia Díaz Blanco
Effects of environmental factors on the gonadal transcriptome of European sea bass (Dicentrarchus labrax), juvenile growth and sex ratios Noelia Díaz Blanco Ph.D. thesis 2014 Submitted in partial fulfillment of the requirements for the Ph.D. degree from the Universitat Pompeu Fabra (UPF). This work has been carried out at the Group of Biology of Reproduction (GBR), at the Department of Renewable Marine Resources of the Institute of Marine Sciences (ICM-CSIC). Thesis supervisor: Dr. Francesc Piferrer Professor d’Investigació Institut de Ciències del Mar (ICM-CSIC) i ii A mis padres A Xavi iii iv Acknowledgements This thesis has been made possible by the support of many people who in one way or another, many times unknowingly, gave me the strength to overcome this "long and winding road". First of all, I would like to thank my supervisor, Dr. Francesc Piferrer, for his patience, guidance and wise advice throughout all this Ph.D. experience. But above all, for the trust he placed on me almost seven years ago when he offered me the opportunity to be part of his team. Thanks also for teaching me how to question always everything, for sharing with me your enthusiasm for science and for giving me the opportunity of learning from you by participating in many projects, collaborations and scientific meetings. I am also thankful to my colleagues (former and present Group of Biology of Reproduction members) for your support and encouragement throughout this journey. To the “exGBRs”, thanks for helping me with my first steps into this world. Working as an undergrad with you Dr. -

Structural Variant Calling by Assembly in Whole Human Genomes: Applications in Hypoplastic Left Heart Syndrome by Matthew Kendzi
STRUCTURAL VARIANT CALLING BY ASSEMBLY IN WHOLE HUMAN GENOMES: APPLICATIONS IN HYPOPLASTIC LEFT HEART SYNDROME BY MATTHEW KENDZIOR THESIS Submitted in partial FulFillment oF tHe requirements for the degree of Master of Science in BioinFormatics witH a concentration in Crop Sciences in the Graduate College of the University oF Illinois at Urbana-Champaign, 2019 Urbana, Illinois Master’s Committee: ProFessor MattHew Hudson, CHair ResearcH Assistant ProFessor Liudmila Mainzer ProFessor SaurabH SinHa ABSTRACT Variant discovery in medical researcH typically involves alignment oF sHort sequencing reads to the human reference genome. SNPs and small indels (variants less than 50 nucleotides) are the most common types oF variants detected From alignments. Structural variation can be more diFFicult to detect From short-read alignments, and thus many software applications aimed at detecting structural variants From short read alignments have been developed. However, these almost all detect the presence of variation in a sample using expected mate-pair distances From read data, making them unable to determine the precise sequence of the variant genome at the speciFied locus. Also, reads from a structural variant allele migHt not even map to the reference, and will thus be lost during variant discovery From read alignment. A variant calling by assembly approacH was used witH tHe soFtware Cortex-var for variant discovery in Hypoplastic Left Heart Syndrome (HLHS). THis method circumvents many of the limitations oF variants called From a reFerence alignment: unmapped reads will be included in a sample’s assembly, and variants up to thousands of nucleotides can be detected, with the Full sample variant allele sequence predicted.