Alternative Splicing in Adhesion- and Motility-Related Genes in Breast Cancer
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Whole-Genome Microarray Detects Deletions and Loss of Heterozygosity of Chromosome 3 Occurring Exclusively in Metastasizing Uveal Melanoma
Anatomy and Pathology Whole-Genome Microarray Detects Deletions and Loss of Heterozygosity of Chromosome 3 Occurring Exclusively in Metastasizing Uveal Melanoma Sarah L. Lake,1 Sarah E. Coupland,1 Azzam F. G. Taktak,2 and Bertil E. Damato3 PURPOSE. To detect deletions and loss of heterozygosity of disease is fatal in 92% of patients within 2 years of diagnosis. chromosome 3 in a rare subset of fatal, disomy 3 uveal mela- Clinical and histopathologic risk factors for UM metastasis noma (UM), undetectable by fluorescence in situ hybridization include large basal tumor diameter (LBD), ciliary body involve- (FISH). ment, epithelioid cytomorphology, extracellular matrix peri- ϩ ETHODS odic acid-Schiff-positive (PAS ) loops, and high mitotic M . Multiplex ligation-dependent probe amplification 3,4 5 (MLPA) with the P027 UM assay was performed on formalin- count. Prescher et al. showed that a nonrandom genetic fixed, paraffin-embedded (FFPE) whole tumor sections from 19 change, monosomy 3, correlates strongly with metastatic death, and the correlation has since been confirmed by several disomy 3 metastasizing UMs. Whole-genome microarray analy- 3,6–10 ses using a single-nucleotide polymorphism microarray (aSNP) groups. Consequently, fluorescence in situ hybridization were performed on frozen tissue samples from four fatal dis- (FISH) detection of chromosome 3 using a centromeric probe omy 3 metastasizing UMs and three disomy 3 tumors with Ͼ5 became routine practice for UM prognostication; however, 5% years’ metastasis-free survival. to 20% of disomy 3 UM patients unexpectedly develop metas- tases.11 Attempts have therefore been made to identify the RESULTS. Two metastasizing UMs that had been classified as minimal region(s) of deletion on chromosome 3.12–15 Despite disomy 3 by FISH analysis of a small tumor sample were found these studies, little progress has been made in defining the key on MLPA analysis to show monosomy 3. -

PLXNB1 (Plexin
Atlas of Genetics and Cytogenetics in Oncology and Haematology OPEN ACCESS JOURNAL AT INIST-CNRS Gene Section Mini Review PLXNB1 (plexin B1) José Javier Gómez-Román, Montserrat Nicolas Martínez, Servando Lazuén Fernández, José Fernando Val-Bernal Department of Anatomical Pathology, Marques de Valdecilla University Hospital, Medical Faculty, University of Cantabria, Santander, Spain (JJGR, MN, SL, JFVB) Published in Atlas Database: March 2009 Online updated version: http://AtlasGeneticsOncology.org/Genes/PLXNB1ID43413ch3p21.html DOI: 10.4267/2042/44702 This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 2.0 France Licence. © 2010 Atlas of Genetics and Cytogenetics in Oncology and Haematology Identity Pseudogene No. Other names: KIAA0407; MGC149167; OTTHUMP00000164806; PLEXIN-B1; PLXN5; SEP Protein HGNC (Hugo): PLXNB1 Location: 3p21.31 Description Local order: The Plexin B1 gene is located between 2135 Amino acids (AA). Plexins are receptors for axon FBXW12 and CCDC51 genes. molecular guidance molecules semaphorins. Plexin signalling is important in pathfinding and patterning of both neurons and developing blood vessels. Plexin-B1 is a surface cell receptor. When it binds to its ligand SEMA4D it activates several pathways by binding of cytoplasmic ligands, like RHOA activation and subsequent changes of the actin cytoskeleton, axon guidance, invasive growth and cell migration. It monomers and heterodimers with PLXNB2 after proteolytic processing. Binds RAC1 that has been activated by GTP binding. It binds PLXNA1 and by similarity ARHGEF11, Note ARHGEF12, ERBB2, MET, MST1R, RND1, NRP1 Size: 26,200 bases. and NRP2. Orientation: minus strand. This family features the C-terminal regions of various plexins. The cytoplasmic region, which has been called DNA/RNA a SEX domain in some members of this family is involved in downstream signalling pathways, by Description interaction with proteins such as Rac1, RhoD, Rnd1 and other plexins. -

Kids First Pediatric Research Program (Kids First) Poster Session at ASHG Accelerating Pediatric Genomics Research Through Collaboration October 15Th, 2019
The Gabriella Miller Kids First Pediatric Research Program (Kids First) Poster Session at ASHG Accelerating Pediatric Genomics Research through Collaboration October 15th, 2019 Background The Gabriella Miller Kids First Pediatric Research Program (Kids First) is a trans- NIH Common Fund program initiated in response to the 2014 Gabriella Miller Kids First Research Act. The program’s vision is to alleviate suffering from childhood cancer and structural birth defects by fostering collaborative research to uncover the etiology of these diseases and support data sharing within the pediatric research community. This is implemented through developing the Gabriella Miller Kids First Data Resource (Kids First Data Resource) and populating this resource with whole genome sequence datasets and associated clinical and phenotypic information. Both childhood cancers and structural birth defects are critical and costly conditions associated with substantial morbidity and mortality. Elucidating the underlying genetic etiology of these diseases has the potential to profoundly improve preventative measures, diagnostics, and therapeutic interventions. Purpose During this evening poster session, attendees will gain a broad understanding of the utility of the genomic data generated by Kids First, learn about the progress of Kids First X01 cohort projects, and observe demonstrations of the tools and functionalities of the recently launched Kids First Data Resource Portal. The session is an opportunity for the scientific community and public to engage with Kids First investigators, collaborators, and a growing community of researchers, patient foundations, and families. Several other NIH and external data efforts will present posters and be available to discuss collaboration opportunities as we work together to accelerate pediatric research. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -
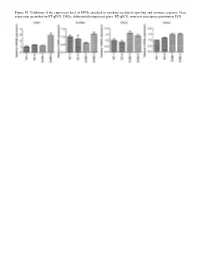
Figure S1. Validation of the Expression Level of Degs Enriched in Cytokine‑Mediated Signaling and Immune Response
Figure S1. Validation of the expression level of DEGs enriched in cytokine‑mediated signaling and immune response. Gene expression quantified by RT‑qPCR. DEGs, differentially expressed genes; RT‑qPCR, reverse‑transcription quantitative PCR. Figure S2. Validation of STAU1‑regulated AS events. IGV‑Sashimi plot revealed (A‑C) three A5SS AS events in three different genes. Reads distribution of each AS event was plotted in the left panel with the transcripts of each gene shown below. The sche‑ matic diagrams depict the structures of two AS events, AS1 (purple line) and AS2 (green line). The exon sequences are denoted by black boxes, the intron sequences by a horizontal line (right panel). RNA‑seq quantification and RT‑qPCR validation of ASEs are presented in the panels on the right. STAU1, double‑stranded RNA‑binding protein Staufen homolog 1; AS, alternative splicing; A5SS, alternative 5'splice site; RNA‑seq, RNA sequencing; RT‑qPCR, reverse‑transcription quantitative PCR. Error bars represent mean ± SEM. *P<0.05. Table SI. Primers used in gene validation experiments. IFIT2‑F CAGCCTACGGCAACTAAA IFIT2‑R GAGCCTTCTCAAAGCACA IFIT3‑F ACACCAAACAATGGCTAC IFIT3‑R TGGACAAACCCTCTAAAC OASL‑F AATGGTGACCGTGATGGG OASL‑R ACCTGAGGATGGAGCAGAG IFI27‑F TTCACTGCGGCGGGAATC IFI27‑R TGGCTGCTATGGAGGACGAG S1PR4‑F TGCTGAAGACGGTGCTGATG S1PR4‑R TGCGGAAGGAGTAGATGATGG CCL5‑F ACGACTGCTGGGTTGGAG CCL5‑R ACCCTGCTGCTTTGCCTA CCL2‑F CTAACCCAGAAACATCCAAT CCL2‑R GCTATGAGCAGCAGGCAC CD44‑F TGGAGGACAGAAAGCCAAGT CD44‑R TTCGCAATGAAACAATCAGTAG PLEKHG2‑M/As‑F CCAAAAGTAAGCCTGTCC PLEKHG2‑M‑R -

Transcriptomic and Proteomic Profiling Provides Insight Into
BASIC RESEARCH www.jasn.org Transcriptomic and Proteomic Profiling Provides Insight into Mesangial Cell Function in IgA Nephropathy † † ‡ Peidi Liu,* Emelie Lassén,* Viji Nair, Celine C. Berthier, Miyuki Suguro, Carina Sihlbom,§ † | † Matthias Kretzler, Christer Betsholtz, ¶ Börje Haraldsson,* Wenjun Ju, Kerstin Ebefors,* and Jenny Nyström* *Department of Physiology, Institute of Neuroscience and Physiology, §Proteomics Core Facility at University of Gothenburg, University of Gothenburg, Gothenburg, Sweden; †Division of Nephrology, Department of Internal Medicine and Department of Computational Medicine and Bioinformatics, University of Michigan, Ann Arbor, Michigan; ‡Division of Molecular Medicine, Aichi Cancer Center Research Institute, Nagoya, Japan; |Department of Immunology, Genetics and Pathology, Uppsala University, Uppsala, Sweden; and ¶Integrated Cardio Metabolic Centre, Karolinska Institutet Novum, Huddinge, Sweden ABSTRACT IgA nephropathy (IgAN), the most common GN worldwide, is characterized by circulating galactose-deficient IgA (gd-IgA) that forms immune complexes. The immune complexes are deposited in the glomerular mesangium, leading to inflammation and loss of renal function, but the complete pathophysiology of the disease is not understood. Using an integrated global transcriptomic and proteomic profiling approach, we investigated the role of the mesangium in the onset and progression of IgAN. Global gene expression was investigated by microarray analysis of the glomerular compartment of renal biopsy specimens from patients with IgAN (n=19) and controls (n=22). Using curated glomerular cell type–specific genes from the published literature, we found differential expression of a much higher percentage of mesangial cell–positive standard genes than podocyte-positive standard genes in IgAN. Principal coordinate analysis of expression data revealed clear separation of patient and control samples on the basis of mesangial but not podocyte cell–positive standard genes. -

3B and Semaphorin-3F in Ovarian Cancer
Published OnlineFirst February 2, 2010; DOI: 10.1158/1535-7163.MCT-09-0664 Research Article Molecular Cancer Therapeutics Hormonal Regulation and Distinct Functions of Semaphorin- 3B and Semaphorin-3F in Ovarian Cancer Doina Joseph1, Shuk-Mei Ho2, and Viqar Syed1 Abstract Semaphorins comprise a family of molecules that influence neuronal growth and guidance. Class-3 sema- phorins, semaphorin-3B (SEMA3B) and semaphorin-3F (SEMA3F), illustrate their effects by forming a com- plex with neuropilins (NP-1 or NP-2) and plexins. We examined the status and regulation of semaphorins and their receptors in human ovarian cancer cells. A significantly reduced expression of SEMA3B (83 kDa), SE- MA3F (90 kDa), and plexin-A3 was observed in ovarian cancer cell lines when compared with normal human ovarian surface epithelial cells. The expression of NP-1, NP-2, and plexin-A1 was not altered in human ovar- ian surface epithelial and ovarian cancer cells. The decreased expression of SEMA3B, SEMA3F, and plexin-A3 was confirmed in stage 3 ovarian tumors. The treatment of ovarian cancer cells with luteinizing hormone, follicle-stimulating hormone, and estrogen induced a significant upregulation of SEMA3B, whereas SEMA3F was upregulated only by estrogen. Cotreatment of cell lines with a hormone and its specific antagonist blocked the effect of the hormone. Ectopic expression of SEMA3B or SEMA3F reduced soft-agar colony for- mation, adhesion, and cell invasion of ovarian cancer cell cultures. Forced expression of SEMA3B, but not SEMA3F, inhibited viability of ovarian cancer cells. Overexpression of SEMA3B and SEMA3F reduced focal adhesion kinase phosphorylation and matrix metalloproteinase-2 and matrix metalloproteinase-9 expression in ovarian cancer cells. -

Downregulation of Rap1gap Through Epigenetic Silencing and Loss of Heterozygosity Promotes Invasion and Progression of Thyroid Tumors
Published OnlineFirst February 2, 2010; DOI: 10.1158/0008-5472.CAN-09-2812 Published Online First on February 2, 2010 as 10.1158/0008-5472.CAN-09-2812 Molecular and Cellular Pathobiology Cancer Research Downregulation of Rap1GAP through Epigenetic Silencing and Loss of Heterozygosity Promotes Invasion and Progression of Thyroid Tumors Hui Zuo1, Manoj Gandhi1, Martin M. Edreira2, Daniel Hochbaum2, Vishwajit L. Nimgaonkar3, Ping Zhang1, James DiPaola1, Viktoria Evdokimova1, Daniel L. Altschuler2, and Yuri E. Nikiforov1 Abstract Thyroid cancer is the most common type of endocrine malignancy, encompassing tumors with various levels of invasive growth and aggressiveness. Rap1GAP, a Rap1 GTPase-activating protein, inhibits the RAS superfamily protein Rap1 by facilitating hydrolysis of GTP to GDP. In this study, we analyzed 197 thyroid tumor samples and showed that Rap1GAP was frequently lost or downregulated in various types of tumors, particularly in the most invasive and aggressive forms of thyroid cancer. The downregulation was due to pro- moter hypermethylation and/or loss of heterozygosity, found in the majority of thyroid tumors. Treatment with demethylating agent 5-aza-deoxycytidine and/or histone deacetylation inhibitor trichostatin A induced gene reexpression in thyroid cells. A genetic polymorphism, Y609C, was seen in 7% of thyroid tumors but was not related to gene downregulation. Loss of Rap1GAP expression correlated with tumor invasiveness but not with specific mutations activating the mitogen-activated protein kinase pathway. Rap1GAP downregulation was required in vitro for cell migration and Matrigel invasion. Recovery of Rap1GAP expression inhibited thy- roid cell proliferation and colony formation. Overall, our findings indicate that epigenetic or genetic loss of Rap1GAP is very common in thyroid cancer, where these events are sufficient to promote cell proliferation and invasion. -

A Set of Regulatory Genes Co-Expressed in Embryonic Human Brain Is Implicated in Disrupted Speech Development
Molecular Psychiatry https://doi.org/10.1038/s41380-018-0020-x ARTICLE A set of regulatory genes co-expressed in embryonic human brain is implicated in disrupted speech development 1 1 1 2 3 Else Eising ● Amaia Carrion-Castillo ● Arianna Vino ● Edythe A. Strand ● Kathy J. Jakielski ● 4,5 6 7 8 9 Thomas S. Scerri ● Michael S. Hildebrand ● Richard Webster ● Alan Ma ● Bernard Mazoyer ● 1,10 4,5 6,11 6,12 13 Clyde Francks ● Melanie Bahlo ● Ingrid E. Scheffer ● Angela T. Morgan ● Lawrence D. Shriberg ● Simon E. Fisher 1,10 Received: 22 September 2017 / Revised: 3 December 2017 / Accepted: 2 January 2018 © The Author(s) 2018. This article is published with open access Abstract Genetic investigations of people with impaired development of spoken language provide windows into key aspects of human biology. Over 15 years after FOXP2 was identified, most speech and language impairments remain unexplained at the molecular level. We sequenced whole genomes of nineteen unrelated individuals diagnosed with childhood apraxia of speech, a rare disorder enriched for causative mutations of large effect. Where DNA was available from unaffected parents, CHD3 SETD1A WDR5 fi 1234567890();,: we discovered de novo mutations, implicating genes, including , and . In other probands, we identi ed novel loss-of-function variants affecting KAT6A, SETBP1, ZFHX4, TNRC6B and MKL2, regulatory genes with links to neurodevelopment. Several of the new candidates interact with each other or with known speech-related genes. Moreover, they show significant clustering within a single co-expression module of genes highly expressed during early human brain development. This study highlights gene regulatory pathways in the developing brain that may contribute to acquisition of proficient speech. -

Genetic Expression and Mutational Profile Analysis in Different
Gao et al. BMC Cancer (2021) 21:786 https://doi.org/10.1186/s12885-021-08442-y RESEARCH Open Access Genetic expression and mutational profile analysis in different pathologic stages of hepatocellular carcinoma patients Xingjie Gao1,2*†, Chunyan Zhao1,2†, Nan Zhang1,2†, Xiaoteng Cui1,2,3, Yuanyuan Ren1,2, Chao Su1,2, Shaoyuan Wu1,2, Zhi Yao1,2 and Jie Yang1,2* Abstract Background: The clinical pathologic stages (stage I, II, III-IV) of hepatocellular carcinoma (HCC) are closely linked to the clinical prognosis of patients. This study aims at investigating the gene expression and mutational profile in different clinical pathologic stages of HCC. Methods: Based on the TCGA-LIHC cohort, we utilized a series of analytical approaches, such as statistical analysis, random forest, decision tree, principal component analysis (PCA), to identify the differential gene expression and mutational profiles. The expression patterns of several targeting genes were also verified by analyzing the Chinese HLivH060PG02 HCC cohort, several GEO datasets, HPA database, and diethylnitrosamine-induced HCC mouse model. Results: We identified a series of targeting genes with copy number variation, which is statistically associated with gene expression. Non-synonymous mutations mainly existed in some genes (e.g.,TTN, TP53, CTNNB1). Nevertheless, no association between gene mutation frequency and pathologic stage distribution was detected. The random forest and decision tree modeling analysis data showed a group of genes related to different HCC pathologic stages, including GAS2L3 and SEMA3F. Additionally, our PCA data indicated several genes associated with different pathologic stages, including SNRPA and SNRPD2. Compared with adjacent normal tissues, we observed a highly expressed level of GAS2L3, SNRPA, and SNRPD2 (P = 0.002) genes in HCC tissues of our HLivH060PG02 cohort. -

Human Induced Pluripotent Stem Cell–Derived Podocytes Mature Into Vascularized Glomeruli Upon Experimental Transplantation
BASIC RESEARCH www.jasn.org Human Induced Pluripotent Stem Cell–Derived Podocytes Mature into Vascularized Glomeruli upon Experimental Transplantation † Sazia Sharmin,* Atsuhiro Taguchi,* Yusuke Kaku,* Yasuhiro Yoshimura,* Tomoko Ohmori,* ‡ † ‡ Tetsushi Sakuma, Masashi Mukoyama, Takashi Yamamoto, Hidetake Kurihara,§ and | Ryuichi Nishinakamura* *Department of Kidney Development, Institute of Molecular Embryology and Genetics, and †Department of Nephrology, Faculty of Life Sciences, Kumamoto University, Kumamoto, Japan; ‡Department of Mathematical and Life Sciences, Graduate School of Science, Hiroshima University, Hiroshima, Japan; §Division of Anatomy, Juntendo University School of Medicine, Tokyo, Japan; and |Japan Science and Technology Agency, CREST, Kumamoto, Japan ABSTRACT Glomerular podocytes express proteins, such as nephrin, that constitute the slit diaphragm, thereby contributing to the filtration process in the kidney. Glomerular development has been analyzed mainly in mice, whereas analysis of human kidney development has been minimal because of limited access to embryonic kidneys. We previously reported the induction of three-dimensional primordial glomeruli from human induced pluripotent stem (iPS) cells. Here, using transcription activator–like effector nuclease-mediated homologous recombination, we generated human iPS cell lines that express green fluorescent protein (GFP) in the NPHS1 locus, which encodes nephrin, and we show that GFP expression facilitated accurate visualization of nephrin-positive podocyte formation in -

Suppl Fig 2 PB2 EUCOMM V3
Maier et al. Supplementary material Semaphorin 4C and 4G are Plexin-B2 ligands required in cerebellar development 2 Supplementary tables 4 Supplementary figures 1 Supplementary Table 1. Exencephaly and postnatal survival of Sema4C mutants (on C57BL/6 background) Genotype Expected frequency Observed frequency Exencephaly Embryonic stages (n=47)* Wild type 25% 25% (12/47) 0% Sema4c+/- 50% 45% (21/47) 0% Sema4c-/- 25% 30% (14/47) 36% (5/14) Postnatal animals (n=417)* Wild type 25% 27% (113/417) Sema4c+/- 50% 61% (253/417) Sema4c-/- 25% 12% (51/417) * Offspring from Sema4c+/- x +/- matings. Embryonic stages scored at E15-E18, postnatal animals scored at P21. 2 Supplementary Table 2. Cerebellar phenotypes of Sema4C and Sema4G mutants (on CD-1 outbred background) Ectopic Fusion of Gap in Normal Gap in IGL granule cells lobules IGL of cerebellum of lobule II in molecular VIII/IX* lobule X Genotype n layer weak strong Wild type 17 100% 0% 0% 0% 0% 0% Sema4c+/- 11 70% 10% 0% 10% 0% 10% Sema4c-/- 10 20% 70% 0% 40% 0% 40% Sema4c+/-; Sema4g+/- 16 55% 18% 0% 18% 0% 18% Sema4c+/-; Sema4g-/- 13 40% 20% 0% 20% 0% 40% Sema4c-/-; Sema4g+/- 10 20% 30% 0% 50% 0% 60% Sema4c-/-; Sema4g-/- 11 0% 55% 0% 55% 55% 82% *Fusions of lobules VIII and IX were scored as “weak” when ectopic granule cells formed a band of cells at the fusion line, and as “strong” when a continuous bridge of granule cells connected the IGL of the two lobules. 3 Supplementary figure legends Maier et al. Suppl. Fig.