111. Fluoroacetates...V. References
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
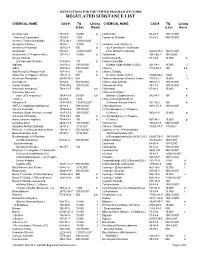
Regulated Substance List
INSTRUCTIONS FOR THE UNIFIED PROGRAM (UP) FORM REGULATED SUBSTANCE LIST CHEMICAL NAME CAS # TQ Listing CHEMICAL NAME CAS # TQ Listing (Lbs) Basis (Lbs) Basis Acetaldehyde 75-07-0 10,000 g Cantharidin 56-25-7 100/10,0001 * Acetone Cyanohydrin 75-86-5 1,000 Carbachol Chloride 51-83-2 500/10,0001 Acetone Thiosemicarbazide 1752-30-3 1,000/10,0001 Acetylene (Ethyne) 74-86-2 10,000 f Carbamic Acid, Methyl-,o- Acrolein (2-Propenal) 107-02-8 500 b (((2,4-Dimethyl-1,3-Dithiolan- Acrylamide 79-06-1 1,000/10,0001 2-YL) Methylene)Amino)- 26419-73-8 100/10,0001 Acrylonitrile (2- Propenenitrile) 107-13-1 10,000 b Carbofuran 1563-66-2 10/10,0001 Acrylyl Chloride Carbon Disulfide 75-15-0 10,000 b (2-Propenoyl Chloride) 814-68-6 100 b Carbon Oxysulfide Aldicarb 116-06-3 100/10,0001 (Carbon Oxide Sulfide (COS)) 463-58-1 10,000 f Aldrin 309-00-2 500/10,0001 Chlorine 7782-50-5 100 a,b Allyl Alcohol (2-Propen-1-ol) 107-18-6 1,000 b Chlorine Dioxide Allylamine (2-Propen-1-Amine) 107-11-9 500 b (Chlorine Oxide (ClO2)) 10049-04-4 1,000 c Aluminum Phosphide 20859-73-8 500 Chlorine Monoxide (Chlorine Oxide) 7791-21-1 10,000 f Aminopterin 54-62-6 500/10,0001 Chlormequat Chloride 999-81-5 100/10,0001 Amiton Oxalate 3734-97-2 100/10,0001 Chloroacetic Acid 79-11-8 100/10,0001 Ammonia, Anhydrous 2 7664-41-7 500 a,b Chloroform 67-66-3 10,000 b Ammonia, Aqueous Chloromethyl Ether (conc 20% or greater) 7664-41-7 20,000 a,b (Methane,Oxybis(chloro-) 542-88-1 100 b * Aniline 62-53-3 1,000 Chloromethyl Methyl Ether Antimycin A 1397-94-0 1,000/10,0001 (Chloromethoxymethane) -

EPCRA.Doc Elizabethtown College
Environmental Affairs EMERGENCY PLANNING AND COMMUNITY RIGHT TO KNOW SCOPE: This policy includes all hazardous materials that are stored and/or used on site and for which there is an MSDS required by OSHA 1910:1200 Hazard Communication. PURPOSE: The purpose of this policy is to establish reporting requirements to provide the public with information on the hazardous chemicals used and or stored at the College. It also is intended to facilitate the development of state and local emergency response plans. PROCESS: ♦ The local emergency planning committee has been notified that the Manager of Human Resources & Safety will serve as the Facility Emergency Coordinator. ♦ The College will provide the Local Emergency Planning Committee and the State Emergency Response Commission with all information needed for emergency planning. ♦ The College will collect and maintain copies of all MSDS(s) required under OSHA 1910:1200 Hazard Communication. The College also subscribes to an MSDS On Demand service so that MSDS(s) will be available at all times. ♦ The College has provided the State Emergency Response Commission, the Local Emergency Planning Committee and the local Fire Department with a complete list of hazardous chemicals used/stored at the College in preference to complete sets of MSDS(s). The College will notify the above agencies of significant changes in the inventory levels of extremely hazardous substances (EHS). ♦ As a matter of practice the College will maintain inventory levels of EHS(s) campus-wide below the Threshold Planning Quantities (TPQ) or 500 lbs, whichever is less, whenever possible. The exception to this will be Sulfuric Acid because of its presence in batteries. -

Downloads/DL Praevention/Fachwissen/Gefahrstoffe/TOXIKOLOGI SCHE BEWERTUNGEN/Bewertungen/Toxbew072-L.Pdf
Distribution Agreement In presenting this thesis or dissertation as a partial fulfillment of the requirements for an advanced degree from Emory University, I hereby grant to Emory University and its agents the non-exclusive license to archive, make accessible, and display my thesis or dissertation in whole or in part in all forms of media, now or hereafter known, including display on the world wide web. I understand that I may select some access restrictions as part of the online submission of this thesis or dissertation. I retain all ownership rights to the copyright of the thesis or dissertation. I also retain the right to use in future works (such as articles or books) all or part of this thesis or dissertation. Signature: _____________________________ ______________ Jedidiah Samuel Snyder Date Statistical analysis of concentration-time extrapolation factors for acute inhalation exposures to hazardous substances By Jedidiah S. Snyder Master of Public Health Global Environmental Health _________________________________________ P. Barry Ryan, Ph.D. Committee Chair _________________________________________ Eugene Demchuk, Ph.D. Committee Member _________________________________________ Paige Tolbert, Ph.D. Committee Member Statistical analysis of concentration-time extrapolation factors for acute inhalation exposures to hazardous substances By Jedidiah S. Snyder Bachelor of Science in Engineering, B.S.E. The University of Iowa 2010 Thesis Committee Chair: P. Barry Ryan, Ph.D. An abstract of A thesis submitted to the Faculty of the Rollins School of Public Health of Emory University in partial fulfillment of the requirements for the degree of Master of Public Health in Global Environmental Health 2015 Abstract Statistical analysis of concentration-time extrapolation factors for acute inhalation exposures to hazardous substances By Jedidiah S. -

Calarp) Program
California Accidental Release Prevention (CalARP) Program Administering Agency Guidance January 31, 2005 Preface This document provides general guidance to help Administering Agencies (AAs) implement and enforce the California Accidental Release Prevention (CalARP) Program. The intent is to identify the elements of the Program applicable to each regulated business, and assist AAs with oversight of the CalARP Program statutes and regulations. This document is not a substitute for the CalARP Program regulations; it does not impose legally binding requirements. About This Document This document follows the format of the California Code of Regulations, Title 19, Division 2, Chapter 4.5: California Accidental Release Prevention (CalARP) Program. The regulatory sections are presented in parentheses for ease of reference. Acknowledgements The California Emergency Management Agency (Cal EMA) would like to thank the following people for their valuable assistance in the preparation of this document: Howard Wines, Hazardous Materials Specialist, City of Bakersfield Fire Department Robert Distaso P.E., Fire Safety Engineer, Orange County Fire Authority Randall L. Sawyer, Supervisor, Accidental Release Prevention Programs, Contra Costa County Health Services Department Beronia Beniamine, Senior Hazardous Materials Specialist, Stanislaus County Environmental Resources Department Angie Proboszcz, Risk Management Program Coordinator, USEPA Region 9 Jon Christenson, Senior Environmental Health Specialist, Merced County Department of Public Health Teresa -
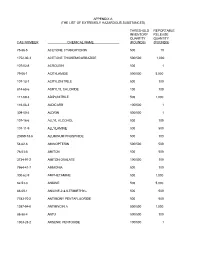
The List of Extremely Hazardous Substances)
APPENDIX A (THE LIST OF EXTREMELY HAZARDOUS SUBSTANCES) THRESHOLD REPORTABLE INVENTORY RELEASE QUANTITY QUANTITY CAS NUMBER CHEMICAL NAME (POUNDS) (POUNDS) 75-86-5 ACETONE CYANOHYDRIN 500 10 1752-30-3 ACETONE THIOSEMICARBAZIDE 500/500 1,000 107-02-8 ACROLEIN 500 1 79-06-1 ACRYLAMIDE 500/500 5,000 107-13-1 ACRYLONITRILE 500 100 814-68-6 ACRYLYL CHLORIDE 100 100 111-69-3 ADIPONITRILE 500 1,000 116-06-3 ALDICARB 100/500 1 309-00-2 ALDRIN 500/500 1 107-18-6 ALLYL ALCOHOL 500 100 107-11-9 ALLYLAMINE 500 500 20859-73-8 ALUMINUM PHOSPHIDE 500 100 54-62-6 AMINOPTERIN 500/500 500 78-53-5 AMITON 500 500 3734-97-2 AMITON OXALATE 100/500 100 7664-41-7 AMMONIA 500 100 300-62-9 AMPHETAMINE 500 1,000 62-53-3 ANILINE 500 5,000 88-05-1 ANILINE,2,4,6-TRIMETHYL- 500 500 7783-70-2 ANTIMONY PENTAFLUORIDE 500 500 1397-94-0 ANTIMYCIN A 500/500 1,000 86-88-4 ANTU 500/500 100 1303-28-2 ARSENIC PENTOXIDE 100/500 1 THRESHOLD REPORTABLE INVENTORY RELEASE QUANTITY QUANTITY CAS NUMBER CHEMICAL NAME (POUNDS) (POUNDS) 1327-53-3 ARSENOUS OXIDE 100/500 1 7784-34-1 ARSENOUS TRICHLORIDE 500 1 7784-42-1 ARSINE 100 100 2642-71-9 AZINPHOS-ETHYL 100/500 100 86-50-0 AZINPHOS-METHYL 10/500 1 98-87-3 BENZAL CHLORIDE 500 5,000 98-16-8 BENZENAMINE, 3-(TRIFLUOROMETHYL)- 500 500 100-14-1 BENZENE, 1-(CHLOROMETHYL)-4-NITRO- 500/500 500 98-05-5 BENZENEARSONIC ACID 10/500 10 3615-21-2 BENZIMIDAZOLE, 4,5-DICHLORO-2-(TRI- 500/500 500 FLUOROMETHYL)- 98-07-7 BENZOTRICHLORIDE 100 10 100-44-7 BENZYL CHLORIDE 500 100 140-29-4 BENZYL CYANIDE 500 500 15271-41-7 BICYCLO[2.2.1]HEPTANE-2-CARBONITRILE,5- -
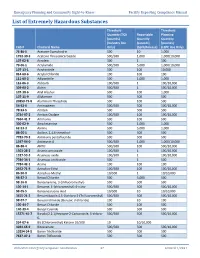
List of Extremely Hazardous Substances
Emergency Planning and Community Right-to-Know Facility Reporting Compliance Manual List of Extremely Hazardous Substances Threshold Threshold Quantity (TQ) Reportable Planning (pounds) Quantity Quantity (Industry Use (pounds) (pounds) CAS # Chemical Name Only) (Spill/Release) (LEPC Use Only) 75-86-5 Acetone Cyanohydrin 500 10 1,000 1752-30-3 Acetone Thiosemicarbazide 500/500 1,000 1,000/10,000 107-02-8 Acrolein 500 1 500 79-06-1 Acrylamide 500/500 5,000 1,000/10,000 107-13-1 Acrylonitrile 500 100 10,000 814-68-6 Acrylyl Chloride 100 100 100 111-69-3 Adiponitrile 500 1,000 1,000 116-06-3 Aldicarb 100/500 1 100/10,000 309-00-2 Aldrin 500/500 1 500/10,000 107-18-6 Allyl Alcohol 500 100 1,000 107-11-9 Allylamine 500 500 500 20859-73-8 Aluminum Phosphide 500 100 500 54-62-6 Aminopterin 500/500 500 500/10,000 78-53-5 Amiton 500 500 500 3734-97-2 Amiton Oxalate 100/500 100 100/10,000 7664-41-7 Ammonia 500 100 500 300-62-9 Amphetamine 500 1,000 1,000 62-53-3 Aniline 500 5,000 1,000 88-05-1 Aniline, 2,4,6-trimethyl- 500 500 500 7783-70-2 Antimony pentafluoride 500 500 500 1397-94-0 Antimycin A 500/500 1,000 1,000/10,000 86-88-4 ANTU 500/500 100 500/10,000 1303-28-2 Arsenic pentoxide 100/500 1 100/10,000 1327-53-3 Arsenous oxide 100/500 1 100/10,000 7784-34-1 Arsenous trichloride 500 1 500 7784-42-1 Arsine 100 100 100 2642-71-9 Azinphos-Ethyl 100/500 100 100/10,000 86-50-0 Azinphos-Methyl 10/500 1 10/10,000 98-87-3 Benzal Chloride 500 5,000 500 98-16-8 Benzenamine, 3-(trifluoromethyl)- 500 500 500 100-14-1 Benzene, 1-(chloromethyl)-4-nitro- 500/500 -

Fluoroacetate Toxicity Hanover, New Hampshire 03755
Gordon W. Gribble' Dartmouth College Fluoroacetate Toxicity Hanover, New Hampshire 03755 Under the stress of World War I1 chemists in England, Table 1. Toxicities of MFA and Other Toxic Compoundso Germany and their allied countries sought to develop chemicals (independently, of course!) which would inca- pacitate, maim, or kill the enemy. These remarkably suc- cessful researches led to the synthesis and large-scale pro- MFA ... 6 duction of several types of warfare agents: nerve gases, FCHzCOINa 0.2 . mustard gas 0.7 . vesicant agents, tear gases, harassing compounds, and, parathion 10 . perhaps the most frightening of all, water poisons. strychnine 5 0.5 For the latter kind of chemical agent it can be easily NaCN ... 10 envisaged that a secret agent could poison the water sup- DFP (nerve agent) . 4 .nlv . of a large" enemv". ~onulace. with hut a small amount of OTaken from several sources. a toxic chemical. The requirements for a water poison are The dose required to kill 50% of the animals by subcutaneous strineent: it should he colorless. odorless. soluble. stable. injection. and rhighly toxic, preferably with a delayed action to pre: Table 2. Selected Physical Properties of MFAa vent early detection. It therefore must have come as quite a surprise to chemists in England, Germany, and Poland Property Value when they discovered independently during the early stag- hoilina ~oint 104' (760 mm) es of the war that a simple derivative of acetic acid fulfills meltingpoint -32' all of the above criteria for an ideal water poison! water solubility 15% This compound is methyl fluoroacetate (MFA) and it, odor faintlv fruitv at 10 DDm along with fluoroacetic acid (FA) and 2-fluoroethanol, represents one of the most toxic classes of non-protein a Reference (4) substances known. -

Selected Analytical Methods for Environmental Remediation and Recovery (SAM) 2017
EPA/600/R-17/356 | September 2017 www.epa.gov/homeland-security-research Selected Analytical Methods for Environmental Remediation and Recovery (SAM) 2017 Office of Research and Development Homeland Security Research Program This page left intentionally blank EPA/600/R-17/356 | September 2017 Selected Analytical Methods for Environmental Remediation and Recovery (SAM) 2017 UNITED STATES ENVIRONMENTAL PROTECTION AGENCY Cincinnati, OH 45268 Office of Research and Development Homeland Security Research Program Disclaimer Disclaimer The U.S. Environmental Protection Agency (EPA) through its Office of Research and Development funded and managed the research described here under Contract EP-C-15-012 to CSRA Inc. This document is undergoing review and has not been approved for publication. The contents reflect the views of the contributors and technical work groups and do not necessarily reflect the views of the Agency. Mention of trade names or commercial products in this document or in the methods referenced in this document does not constitute endorsement or recommendation for use. Questions concerning this document or its application should be addressed to: Romy Campisano National Homeland Security Research Center Office of Research and Development (NG16) U.S. Environmental Protection Agency 26 West Martin Luther King Drive Cincinnati, OH 45268 (513) 569-7016 [email protected] Kathy Hall National Homeland Security Research Center Office of Research and Development (NG16) U.S. Environmental Protection Agency 26 West Martin Luther King -

List of Lists
United States Office of Solid Waste EPA 550-B-10-001 Environmental Protection and Emergency Response May 2010 Agency www.epa.gov/emergencies LIST OF LISTS Consolidated List of Chemicals Subject to the Emergency Planning and Community Right- To-Know Act (EPCRA), Comprehensive Environmental Response, Compensation and Liability Act (CERCLA) and Section 112(r) of the Clean Air Act • EPCRA Section 302 Extremely Hazardous Substances • CERCLA Hazardous Substances • EPCRA Section 313 Toxic Chemicals • CAA 112(r) Regulated Chemicals For Accidental Release Prevention Office of Emergency Management This page intentionally left blank. TABLE OF CONTENTS Page Introduction................................................................................................................................................ i List of Lists – Conslidated List of Chemicals (by CAS #) Subject to the Emergency Planning and Community Right-to-Know Act (EPCRA), Comprehensive Environmental Response, Compensation and Liability Act (CERCLA) and Section 112(r) of the Clean Air Act ................................................. 1 Appendix A: Alphabetical Listing of Consolidated List ..................................................................... A-1 Appendix B: Radionuclides Listed Under CERCLA .......................................................................... B-1 Appendix C: RCRA Waste Streams and Unlisted Hazardous Wastes................................................ C-1 This page intentionally left blank. LIST OF LISTS Consolidated List of Chemicals -

(A) Pupil Sizes. Left Eye Exposed to Di-/Wpropyl Phosphorofluoi Idate (0
(a) Hot* * (*>) Pupil sizes. Left eye exposed to di-/wpropyl phosphorofluoi idate (0-008 mg./l. 2 min. exposure): (a) 3 hr. after exposure; (b) '24 hr. after exj)osure. SOME ASPECTS OF THE CHEMISTRY AND TOXIC ACTION OF ORGANIC COMPOUNDS CONTAINING PHOSPHORUS AND FLUORINE BY BERNARD CHARLES SAUNDERS M.A., PH.D., Sc.D., D.Sc., F.R.I.C. Fellow and Charles Kingsley Lecturer in Natural Sciences, Magdalene College, University Lecturer in Chemistry, Cambridge WITH A FOREWORD BY PROFESSOR SIR ALEXANDER TODD M.A., D.Sc., LL.D., F.R.S. Professor of Organic Chemistry, Cambridge CAMBRIDGE AT THE UNIVERSITY PRESS 1957 63092 PUBLISHED BY THE SYNDICS OF THE CAMBRIDGE UNIVERSITY PRESS Bentley House, 200 Euston Road, London, N.W. I American Branch: 32 East 57th Street, New York 22, N.Y. By the same author NOTES ON QUALITATIVE ANALYSIS Printed in Great Britain at the University Press, Cambridge (Brooke Crutchley, University Printer) CONTENTS FOBEWORD page xiii PREFACE xv CHAPTER I. INTRODUCTION AND GENERAL SURVEY 1 Phosphorofluoridates 1 Production on a technical scale 6 Phosphorodiamidic fluorides 7 Fluoroacetates 10 2-Fluoroethyl fluoroacetate 12 Other compounds 15 Fluoro-aspirin, £>. 15. Di-2-nuoroethylphosphoronuorid- ate, p. 15. Triethyl-lead fluoroacetate, p. 16. 'Sesqui- fluoro-H', p. 16. Fluorine-containing ammonium salts, p. M Particular applications 18 CHAPTER II. NOMENCLATURE OF ESTERS CON- TAINING PHOSPHORUS 21 Early nomenclature 21 Notes 23 Accepted nomenclature 25 Notes on the agreed system for compounds con- taining only one phosphorus atom 27 CHAPTER III. NOTES ON THE MAMMALIAN NER- VOUS SYSTEM 28 The transmission of nervous effects 28 The autonomic nervous system 31 The sympathetic nervous system, p. -

Some Toxicological Aspects of Protection Forestry : a Treatise
- A TREATISE - Protection Fore Branch No. 93 Copy No.: Date: November 1970 Forest Research Institute, Protection Fore Branch, RANGIORA. 1 .. "I have yet to see any problem, however complicated, which, when. you looked at it the right way, did not become more complicated.," Paul Alderson 2 .. CONTENTS PAGE PREFACE 3-5 INTRODUCTION 6 CHAPTER ONE 7-20 SODiill1 MONOFLUOROACETATE - COMPOUND 1080 CHAPTER TWO 21-37 DERIVATIVES OF SODIUM FLUOROACETATE CHAPTER THREE 38-45 SYSTEMIC TOXICITY AND TRANSLOCATION CHAPTER FOUR 46-48 REPRODUCTION INHIBITORS AND CHEMOSTERILANTS CHAPTER FIVE 49-53 ANALYTICAL PRCOEDURES REFERENCES 54-57 POSTSCRIPT PREFACE Toxicology, the study of toxins and poisons, is today a multi disciplinary system of knowledge which acquires and inter-relates information from many branches of science. Within the sphere of Protection Forestry, toxicology is concerned not only with biochemical and physiological derangements, but also with their ecological responses in animal and plant communities. Thus, it represents an important aspect of biochemical ecology. To those acquainted with the problems of wildlife control under the present policy of Protection Forestry it has long been obvious that, in any one of the diverse control programmes, much additional information and benefit could be obtained from a more precise definition and a clearer recognition of the toxicological aspects that form the basis of the majority of these programmes. Thus, the responsibilities of a toxicologist confronted with these problems are many-fold. Not only is he required to select, from the background of his in the various disciplines, a specific approach to a specific problem 9 but also, he must be able to inculcate in other investigators an understanding, or at least an appreciation, of the reasons for this specificity. -

Environmental Protection Agency Pt. 355, App. B
Environmental Protection Agency Pt. 355, App. B [Alphabetical Order] Reportable Threshold plan- CAS No. Chemical name Notes quantity * ning quantity (pounds) (pounds) 5344–82–1 ............ Thiourea, (2-Chlorophenyl)- ....................................... ..................... 100 100/10,000 614–78–8 .............. Thiourea, (2-Methylphenyl)- ....................................... ..................... 500 500/10,000 7550–45–0 ............ Titanium Tetrachloride ................................................ ..................... 1,000 100 584–84–9 .............. Toluene 2,4-Diisocyanate ........................................... ..................... 100 500 91–08–7 ................ Toluene 2,6-Diisocyanate ........................................... ..................... 100 100 110–57–6 .............. Trans-1,4-Dichlorobutene ........................................... ..................... 500 500 1031–47–6 ............ Triamiphos .................................................................. ..................... 500 500/10,000 24017–47–8 .......... Triazofos ..................................................................... ..................... 500 500 76–02–8 ................ Trichloroacetyl Chloride .............................................. ..................... 500 500 115–21–9 .............. Trichloroethylsilane ..................................................... d .................. 500 500 327–98–0 .............. Trichloronate ............................................................... e .................