Duchenne and Becker Muscular Dystrophy: Clinical Features Genetics, and Diagnostics
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Mcleod Neuroacanthocytosis Syndrome
NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health. Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993- 2017. McLeod Neuroacanthocytosis Syndrome Hans H Jung, MD Department of Neurology University Hospital Zurich Zurich, Switzerland [email protected] Adrian Danek, MD Neurologische Klinik Ludwig-Maximilians-Universität München, Germany ed.uml@kenad Ruth H Walker, MD, MBBS, PhD Department of Neurology Veterans Affairs Medical Center Bronx, New York [email protected] Beat M Frey, MD Blood Transfusion Service Swiss Red Cross Schlieren/Zürich, Switzerland [email protected] Christoph Gassner, PhD Blood Transfusion Service Swiss Red Cross Schlieren/Zürich, Switzerland [email protected] Initial Posting: December 3, 2004; Last Update: May 17, 2012. Summary Clinical characteristics. McLeod neuroacanthocytosis syndrome (designated as MLS throughout this review) is a multisystem disorder with central nervous system (CNS), neuromuscular, and hematologic manifestations in males. CNS manifestations are a neurodegenerative basal ganglia disease including (1) movement disorders, (2) cognitive alterations, and (3) psychiatric symptoms. Neuromuscular manifestations include a (mostly subclinical) sensorimotor axonopathy and muscle weakness or atrophy of different degrees. Hematologically, MLS is defined as a specific blood group phenotype (named after the first proband, Hugh McLeod) that results from absent expression of the Kx erythrocyte antigen and weakened expression of Kell blood group antigens. The hematologic manifestations are red blood cell acanthocytosis and compensated hemolysis. Allo-antibodies in the Kell and Kx blood group system can cause strong reactions to transfusions of incompatible blood and severe anemia in newborns of Kell-negative mothers. -

Peripheral Neuropathy in Complex Inherited Diseases: an Approach To
PERIPHERAL NEUROPATHY IN COMPLEX INHERITED DISEASES: AN APPROACH TO DIAGNOSIS Rossor AM1*, Carr AS1*, Devine H1, Chandrashekar H2, Pelayo-Negro AL1, Pareyson D3, Shy ME4, Scherer SS5, Reilly MM1. 1. MRC Centre for Neuromuscular Diseases, UCL Institute of Neurology and National Hospital for Neurology and Neurosurgery, London, WC1N 3BG, UK. 2. Lysholm Department of Neuroradiology, National Hospital for Neurology and Neurosurgery, London, WC1N 3BG, UK. 3. Unit of Neurological Rare Diseases of Adulthood, Carlo Besta Neurological Institute IRCCS Foundation, Milan, Italy. 4. Department of Neurology, University of Iowa, 200 Hawkins Drive, Iowa City, IA 52242, USA 5. Department of Neurology, University of Pennsylvania, Philadelphia, PA 19014, USA. * These authors contributed equally to this work Corresponding author: Mary M Reilly Address: MRC Centre for Neuromuscular Diseases, 8-11 Queen Square, London, WC1N 3BG, UK. Email: [email protected] Telephone: 0044 (0) 203 456 7890 Word count: 4825 ABSTRACT Peripheral neuropathy is a common finding in patients with complex inherited neurological diseases and may be subclinical or a major component of the phenotype. This review aims to provide a clinical approach to the diagnosis of this complex group of patients by addressing key questions including the predominant neurological syndrome associated with the neuropathy e.g. spasticity, the type of neuropathy, and the other neurological and non- neurological features of the syndrome. Priority is given to the diagnosis of treatable conditions. Using this approach, we associated neuropathy with one of three major syndromic categories - 1) ataxia, 2) spasticity, and 3) global neurodevelopmental impairment. Syndromes that do not fall easily into one of these three categories can be grouped according to the predominant system involved in addition to the neuropathy e.g. -

Assessment of a Targeted Gene Panel for Identification of Genes Associated with Movement Disorders
Supplementary Online Content Montaut S, Tranchant C, Drouot N, et al; French Parkinson’s and Movement Disorders Consortium. Assessment of a targeted gene panel for identification of genes associated with movement disorders. JAMA Neurol. Published online June 18, 2018. doi:10.1001/jamaneurol.2018.1478 eMethods. Supplemental methods. eTable 1. Name, phenotype and inheritance of the genes included in the panel. eTable 2. Probable pathogenic variants identified in a cohort of 23 patients with cerebellar ataxia using WES analysis. eTable 3. Negative cases in a cohort of 23 patients with cerebellar ataxia studied using WES analysis. eTable 4. Variants of unknown significance (VUSs) identified in the cohort. eFigure 1. Examples of pedigrees of cases with identified causative variants. eFigure 2. Pedigrees suggesting mendelian inheritance in negative cases. eFigure 3. Examples of pedigrees of cases with identified VUSs. eResults. Supplemental results. This supplementary material has been provided by the authors to give readers additional information about their work. © 2018 American Medical Association. All rights reserved. Downloaded From: https://jamanetwork.com/ on 10/02/2021 eMethods. Supplemental methods Patients selection In the multicentric, prospective study, patients were selected from 25 French, 1 Luxembourg and 1 Algerian tertiary MDs centers between September 2014 and July 2016. Inclusion criteria were patients (1) who had developed one or several chronic MDs (2) with an age of onset below 40 years and/or presence of a family history of MDs. Patients suffering from essential tremor, tic or Gilles de la Tourette syndrome, pure cerebellar ataxia or with clinical/paraclinical findings suggestive of an acquired cause were excluded. -

Ruth H. Walker, MB., Ch.B., Ph.D. Departments of Neurology, James J
A flow chart for the evaluation of chorea Ruth H. Walker, MB., Ch.B., Ph.D. Departments of Neurology, James J. Peters Veterans Affairs Medical Center, Bronx, NY, and Mount Sinai School of Medicine, New York, NY [email protected] Patient with chorea Streptococcal? Age of Infant/child +ve Autosomal recessive/sporadic sore throat, rheumatic Sydenham's chorea Onset? heart disease ASO, anti-DNAse Lesch-Nyhan syndrome B titers Confirm with gene test Tardive dyskinesia Autosomal Adult X-linked Inheritance? Normal recessive Delayed Autosomal Medication- Yes Direct Check Yes Childhood onset, dominant Time course? gouty arthritis, MRI; normal induced? Immediate or side effect uric acid self-mutilation? Fe in basal dose related Calcium Acute infarct in ganglia ? deposition posterior limb of in basal internal capsule. ganglia. Diffusion- No No Pantothenate Phospholipase-associated CT scan weighted MRI Normal neurodegeneration kinase-associated Yes No Infantile bilateral neurodegeneration striatal necrosis •Mix blood 1:1 with 0.9% NaCl containing 10IU/ml heparin Yes Structural lesion; •Incubate at room temperature for 30-120 min on a shaker •Childhood onset PKAN: T2 weighted MRI Stroke, tumor •Take 5+ microphotographs from each wet preparation •Occasional later onset Yes Leigh’s syndrome, (phase-contrast microscope) •Pigmentary retinopathy MRI arteriovenous malformation, Lactate/ •Count cells with spicules: normal value < 6.3 % •Dystonia PANK2 Normal PLA2G6 other mitochrondial? Lubag •10% have acanthocytosis Ceruloplasmin? calcification (IBG1?), etc pyruvate No No Yes mutation? No mutation? Confirm with gene test Acanthocytosis? Filipino? Confirm with gene test Basal ganglia No No Typical phenotype = dystonia- necrosis? parkinsonism, but should be Other neurodegeneration considered in any Filipino with an Yes with brain iron unexplained movement disorder, Caudate Yes including women Yes Recent Post-infectious Accumulation disorder… nucleus FAHN, MPAN, BPAN… Yes infection? striatal necrosis •Full panel of 23 Kell abs usually available atrophy? Courtesy of Dr Hans H. -

(12) Patent Application Publication (10) Pub. No.: US 2010/0210567 A1 Bevec (43) Pub
US 2010O2.10567A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2010/0210567 A1 Bevec (43) Pub. Date: Aug. 19, 2010 (54) USE OF ATUFTSINASATHERAPEUTIC Publication Classification AGENT (51) Int. Cl. A638/07 (2006.01) (76) Inventor: Dorian Bevec, Germering (DE) C07K 5/103 (2006.01) A6IP35/00 (2006.01) Correspondence Address: A6IPL/I6 (2006.01) WINSTEAD PC A6IP3L/20 (2006.01) i. 2O1 US (52) U.S. Cl. ........................................... 514/18: 530/330 9 (US) (57) ABSTRACT (21) Appl. No.: 12/677,311 The present invention is directed to the use of the peptide compound Thr-Lys-Pro-Arg-OH as a therapeutic agent for (22) PCT Filed: Sep. 9, 2008 the prophylaxis and/or treatment of cancer, autoimmune dis eases, fibrotic diseases, inflammatory diseases, neurodegen (86). PCT No.: PCT/EP2008/007470 erative diseases, infectious diseases, lung diseases, heart and vascular diseases and metabolic diseases. Moreover the S371 (c)(1), present invention relates to pharmaceutical compositions (2), (4) Date: Mar. 10, 2010 preferably inform of a lyophilisate or liquid buffersolution or artificial mother milk formulation or mother milk substitute (30) Foreign Application Priority Data containing the peptide Thr-Lys-Pro-Arg-OH optionally together with at least one pharmaceutically acceptable car Sep. 11, 2007 (EP) .................................. O7017754.8 rier, cryoprotectant, lyoprotectant, excipient and/or diluent. US 2010/0210567 A1 Aug. 19, 2010 USE OF ATUFTSNASATHERAPEUTIC ment of Hepatitis BVirus infection, diseases caused by Hepa AGENT titis B Virus infection, acute hepatitis, chronic hepatitis, full minant liver failure, liver cirrhosis, cancer associated with Hepatitis B Virus infection. 0001. The present invention is directed to the use of the Cancer, Tumors, Proliferative Diseases, Malignancies and peptide compound Thr-Lys-Pro-Arg-OH (Tuftsin) as a thera their Metastases peutic agent for the prophylaxis and/or treatment of cancer, 0008. -

Contiguous Gene Deletion of Chromosome Xp in Three Families
Case Report iMedPub Journals Journal of Rare Disorders: Diagnosis & Therapy 2015 http://wwwimedpub.com ISSN 2380-7245 Vol. 1 No. 1:3 DOI: 10.21767/2380-7245.100003 Contiguous Gene Deletion of Shailly Jain-Ghai1,5, Stephanie Skinner1, Chromosome Xp in Three Families Jessica Hartley2,3, Encompassing OTC, RPGR and Stephanie Fox4, TSPAN7 Genes Daniela Buhas4, Cheryl Rockman- Greenberg2,3 and Alicia Chan1,5 Abstract Ornithine transcarbamylase deficiency (OTCD) is the most common urea cycle 1 Medical Genetics Clinic, Stollery disorder. The classic presentation in males is hyperammonemic encephalopathy Children's Hospital, Edmonton, Alberta, in the early neonatal period. Given the X-linked inheritance of OTCD, presentation Canada in females is highly variable. We present three families with different contiguous 2 Program in Genetics and Metabolism, Winnipeg Regional Health Authority gene deletions on chromosome Xp. Deletion ofRPGR , OTC and TSPAN7 is common and University of Manitoba, Winnipeg, to all three families in our series. These cases highlight the variable phenotype Manitoba, Canada in manifesting OTCD female carriers, the complexity of OTCD management and 3 Department of Biochemistry and complex issues surrounding the option of liver transplantation when multiple Medical Genetics, University of other genetic factors play a role. Manitoba, Winnipeg, Manitoba, Canada 4 Department of Medical Genetics, Keywords: Ornithine transcarbamylase; Ornithine transcarbamylase deficiency; Montreal Children’s Hospital, McGill Contiguous gene deletion; -

Wo 2009/033784 A2
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (43) International Publication Date (10) International Publication Number 19 March 2009 (19.03.2009) PCT WO 2009/033784 A2 (51) International Patent Classification: (74) Agent: ARTH, Hans-Lothar; ABK Patent Attorneys, Jas- A61K 38/17 (2006.01) A61P 11/00 (2006.01) minweg 9, 14052 Berlin (DE). A61K 38/01 (2006.01) A61P 25/28 (2006.01) A61P 31/18 (2006.01) A61P 31/00 (2006.01) (81) Designated States (unless otherwise indicated, for every A61P 3/00 (2006.01) A61P 35/00 (2006.01) kind of national protection available): AE, AG, AL, AM, A61P 9/00 (2006.01) A61P 37/00 (2006.01) AO, AT,AU, AZ, BA, BB, BG, BH, BR, BW, BY, BZ, CA, CH, CN, CO, CR, CU, CZ, DE, DK, DM, DO, DZ, EC, EE, (21) International Application Number: EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, HR, HU, ID, PCT/EP2008/007968 IL, IN, IS, JP, KE, KG, KM, KN, KP, KR, KZ, LA, LC, LK, LR, LS, LT, LU, LY, MA, MD, ME, MG, MK, MN, MW, (22) International Filing Date: MX, MY, MZ, NA, NG, NI, NO, NZ, OM, PG, PH, PL, PT, 9 September 2008 (09.09.2008) RO, RS, RU, SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TJ, TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, (25) Filing Language: English ZW (26) Publication Language: English (84) Designated States (unless otherwise indicated, for every kind of regional protection available): ARIPO (BW, GH, (30) Priority Data: GM, KE, LS, MW, MZ, NA, SD, SL, SZ, TZ, UG, ZM, 07017766.2 11 September 2007 (11.09.2007) EP ZW), Eurasian (AM, AZ, BY, KG, KZ, MD, RU, TJ, TM), European (AT,BE, BG, CH, CY, CZ, DE, DK, EE, ES, FI, (71) Applicant (for all designated States except US): MONDO- FR, GB, GR, HR, HU, IE, IS, IT, LT,LU, LV,MC, MT, NL, BIOTECH LABORATORIES AG [LLLI]; Herrengasse NO, PL, PT, RO, SE, SI, SK, TR), OAPI (BF, BJ, CF, CG, 21, FL-9490 Vaduz (LI). -

Unusual Muscle Pathology in Mcleod Syndrome
J Neurol Neurosurg Psychiatry 2000;69:655–657 655 J Neurol Neurosurg Psychiatry: first published as 10.1136/jnnp.69.5.655 on 1 November 2000. Downloaded from SHORT REPORT Unusual muscle pathology in McLeod syndrome M H Barnett, F Yang, H Iland, J D Pollard Abstract antigens, and the absence of the Kx antigen.4 Muscle pathology in McLeod syndrome is The responsible gene, XK, is located on chro- usually mild; patchy necrotic or regener- mosome Xp21, between the loci of chronic ating fibres, occasional internal nuclei, granulomatous disease and Duchenne muscu- and the absence of an inflammatory cell lar dystrophy5; rarely, these disorders may infiltrate are the usual findings. We report occur concurrently.67The muscle biopsy find- on a 29 year old man presenting with ings are variable, but usually subtle.2 We report chronic fatiguability and excessive sweat- on a patient with the McLeod phenotype and ing in whom an open quadriceps muscle striking atypical mononuclear cell infiltrates on biopsy demonstrated grouped necrotic muscle biopsy. fibres accompanied by striking patchy mononuclear cell infiltrates. The diagno- Case report sis of McLeod syndrome was made on the A 29 year old carpenter presented witha5year basis of red blood cell acanthocytosis, history of persistent fatigue and excessive raised serum creatine kinase, and weak sweating on minimal exertion, initially attrib- expression of Kell blood group antigens. uted to a febrile illness diagnosed as infectious The quadriceps muscle infiltrate con- mononucleosis. There were no classic night sisted principally of histologically typical sweats or weight loss, nor any myalgias or defi- copyright. macrophages. -

Download CGT Exome V2.0
CGT Exome version 2. -
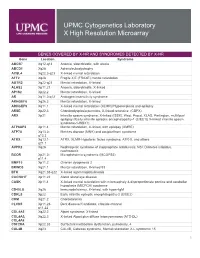
Genes Covered and Disorders Detected by X-HR Microarray
UPMC Cytogenetics Laboratory X High Resolution Microarray GENES COVERED BY X-HR AND SYNDROMES DETECTED BY X-HR Gene Location Syndrome ABCB7 Xq12-q13 Anemia, sideroblastic, with ataxia ABCD1 Xq28 Adrenoleukodystrophy ACSL4 Xq22.3-q23 X-linked mental retardation AFF2 Xq28 Fragile X E (FRAXE) mental retardation AGTR2 Xq22-q23 Mental retardation, X-linked ALAS2 Xp11.21 Anemia, sideroblastic, X-linked AP1S2 Xp22.2 Mental retardation, X-linked AR Xq11.2-q12 Androgen insensitivity syndrome ARHGEF6 Xq26.3 Mental retardation, X-linked ARHGEF9 Xq11.1 X-linked mental retardation (XLMR)//Hyperekplexia and epilepsy ARSE Xp22.3 Chondrodysplasia punctata, X-linked recessive (CDPX) ARX Xp21 Infantile spasm syndrome, X-linked (ISSX), West, Proud, XLAG, Partington, multifocal epilepsy //Early infantile epileptic encephalopathy-1 (EIEE1)( X-linked infantile spasm syndrome-1-ISSX1) ATP6AP2 Xp11.4 Mental retardation, X-linked, with epilepsy (XMRE) ATP7A Xq13.2- Menkes disease (MNK) and occipital horn syndrome q13.3 ATRX Xq13.1- ATRX, XLMR-Hypotonic facies syndrome, ATR-X, and others q21.1 AVPR2 Xq28 Nephrogenic syndrome of inappropriate antidiuresis; NSI; Diabetes insipidus, nephrogenic BCOR Xp21.2- Microphthalmia syndromic (MCOPS2) p11.4 BMP15 Xp11.2 Ovarian dysgenesis 2 BRWD3 Xq21.1 Mental retardation, X-linked 93 BTK Xq21.33-q22 X-linked agammaglobulinemia CACNA1F Xp11.23 Aland Island eye disease CASK Xp11.4 X-linked mental retardation with microcephaly & disproportionate pontine and cerebellar hypoplasia (MICPCH) syndrome CD40LG Xq26 Immunodeficiency, -

Female Carriers of Duchenne Muscular Dystrophy
Review Article Journal of J Genet Med 2013;10(2):94-98 JGM Genetic Medicine http://dx.doi.org/10.5734/JGM.2013.10.2.94 ISSN 1226-1769 (Print) 2233-9108 (Online) Female Carriers of Duchenne Muscular Dystrophy Yu Na Cho and Young-Chul Choi* Department of Neurology, Gangnam Severance Hospital, Yonsei University College of Medicine, Seoul, Korea Dystrophinopathy, caused by mutations in the DMD gene, presents with variable clinical phenotypes ranging from the severe Duchenne muscular dystrophy (DMD) to the milder Becker muscular dystrophy(BMD) forms. DMD is a recessive X-linked form of muscular dystrophy. Two-thirds of mothers of affected males are thought to be DMD carriers. Approximately 2.5-7.8% of female DMD carriers have muscle weakness and are categorized as manifesting DMD carriers. The symptoms of female carriers of DMD range from mild muscle weakness to severe gait problems. The most commonly presented symptom is mild proximal muscle weakness, which is often asymmetric and progressive, but shows variable clinical spectrum with BMD of more severe DMD-like phenotype. Atypical presentations in manifesting carriers are myalgia or cramps without limb weakness, isolated cardiomyopathy and camptocormia. Multiplex PCR and MLPA analysis are common techniques to identify mutations in the DMD gene. Relationship between X-chromosome inactivation and clinical severity is not clear. Female carriers of DMD are not less common, and they have an important role of birth of a male DMD. Key words: Dystrophinopathy, Duchenne muscular dystrophy, Female carrier, Multiplex ligation-dependent probe amplification Introduction males are thought to be DMD carriers. Approximately 2.5-7.8% of female DMD carriers have muscle weakness and are categorized as manifesting DMD carriers.1, 2, 4) Therefore, here we reviewed Duchenne muscular dystrophy (DMD) is a recessive X-linked recent studies to meet the need for a better understanding about form of muscular dystrophy. -

Mcleod Neuroacanthocytosis Syndrome
McLeod neuroacanthocytosis syndrome Description McLeod neuroacanthocytosis syndrome is primarily a neurological disorder that occurs almost exclusively in boys and men. This disorder affects movement in many parts of the body. People with McLeod neuroacanthocytosis syndrome also have abnormal star- shaped red blood cells (acanthocytosis). This condition is one of a group of disorders called neuroacanthocytoses that involve neurological problems and abnormal red blood cells. McLeod neuroacanthocytosis syndrome affects the brain and spinal cord (central nervous system). Affected individuals have involuntary movements, including jerking motions (chorea), particularly of the arms and legs, and muscle tensing (dystonia) in the face and throat, which can cause grimacing and vocal tics (such as grunting and clicking noises). Dystonia of the tongue can lead to swallowing difficulties. Seizures occur in approximately half of all people with McLeod neuroacanthocytosis syndrome. Individuals with this condition may develop difficulty processing, learning, and remembering information (cognitive impairment). They may also develop psychiatric disorders, such as depression, bipolar disorder, psychosis, or obsessive-compulsive disorder. People with McLeod neuroacanthocytosis syndrome also have problems with their muscles, including muscle weakness (myopathy) and muscle degeneration (atrophy). Sometimes, nerves that connect to muscles atrophy (neurogenic atrophy), leading to loss of muscle mass and impaired movement. Individuals with McLeod neuroacanthocytosis