Oxidation of Functionally Substituted Carbanions Alan Greenway Bemis Iowa State University
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

(12) Patent Application Publication (10) Pub. No.: US 2014/0046022 A1 Takahashi (43) Pub
US 20140046022A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2014/0046022 A1 Takahashi (43) Pub. Date: Feb. 13, 2014 (54) FLUORENE COMPOUND (30) Foreign Application Priority Data (71) Applicant: Ajinomoto Co., Inc., Tokyo (JP) Mar. 12, 2009 (JP) ................................. 2009-06O291 (72) Inventor: Daisuke Takahashi, Mie (JP) Publication Classification (73) Assignee: Ajinomoto Co., Inc., Tokyo (JP) (51) Int. Cl. C07K L/06 (2006.01) (21) Appl. No.: 14/027,961 CD7C 43/21 (2006.01) (52) U.S. Cl. (22) Filed: Sep. 16, 2013 CPC ................. C07K I/062 (2013.01); C07C 43/21 (2013.01) Related U.S. Application Data USPC ........................................... 530/335:568/634 (62) Division of application No. 12/723,027, filed on Mar. (57) ABSTRACT 12, 2010, now Pat. No. 8,569,453. Particular compounds having a fluorene skeleton are Superior (60) Provisional application No. 61/159,998, filed on Mar. in broad utility and stability, as a protecting reagent for liquid 13, 2009. phase synthesis of amino acids and/or peptides. US 2014/0046022 A1 Feb. 13, 2014 FLUORENE COMPOUND carrier, an isolation target compound can be selectively pre cipitated from a homogeneous solution state, in other words, CROSS REFERENCES TO RELATED a particular compound can be isolated after a liquid phase APPLICATIONS reaction when other soluble components still remain in a Solution, thus obviating the need to consider extraction and 0001. This application is a division of U.S. Ser. No. precipitation conditions for each compound. 12/723,027, filed Mar. 12, 2010, and claims priority to U.S. 0010. However, when a polymer is used as a carrier mol Provisional Patent Applications No. -

Exam 1 (February 23, 2004) ID# ______
Chemistry 211 Name ___________________________ Exam 1 (February 23, 2004) ID# ___________________________ 10 1. You desire to synthesize 3-ethyl-3-pentanol starting with an ester. (i) What would be the name of the ester, and what is the name for the Grignard reagent (e.g., methyl magnesium bromide)? (ii) For the carbons shown in the product, show plausible hydrocarbons that you could start with to produce the ester and the Grignard reagent (as in a retrosynthesis). 12 2. (i) Show the step-by-step process required to produce propyllithium, which requires a free radical reaction mechanism, . (ii) Show the complete reaction mechanism for reaction between propyllithium and the correct ketone to produce 3-propyl-3-pentanol. (iii) Propose a possible reaction mechanism by which dipropyl cuprate (Cu+ with two propyl groups attached) could react with ethyl bromide to produce a new hydrocarbon. (This is a thinking exercise! So, think! () 8 3. As mentioned in the text, diethyl ether, pentane, and 1-butanol have similar molar masses, but different physical properties. Boiling points are 35oC, 36oC, and 117oC, respectively. Their respective solubilities in water are 7.5g/100mL, insoluble, and 9g/100mL. (i) Draw structures for each of these compounds. (ii) Justify the observed boiling points and their solubilities. 16 4. Draw structures of the following compounds 2,3-heptanediol isopropyllithium benzylmagnesium bromide benzoic acid benzaldehylde dimethyl sulfide t-butyl methanoate dibutyl ketone 12 5. Alcohols can be oxidized to produce other compounds, and can be produced by reduction. For the reactions shown below, show the structure for the expected product (if reaction does not occur, state: No Reaction) when treated with the indicated oxidizing or reducing agents. -

Polycyclic Aromatic Hydrocarbon Structure Index
NIST Special Publication 922 Polycyclic Aromatic Hydrocarbon Structure Index Lane C. Sander and Stephen A. Wise Chemical Science and Technology Laboratory National Institute of Standards and Technology Gaithersburg, MD 20899-0001 December 1997 revised August 2020 U.S. Department of Commerce William M. Daley, Secretary Technology Administration Gary R. Bachula, Acting Under Secretary for Technology National Institute of Standards and Technology Raymond G. Kammer, Director Polycyclic Aromatic Hydrocarbon Structure Index Lane C. Sander and Stephen A. Wise Chemical Science and Technology Laboratory National Institute of Standards and Technology Gaithersburg, MD 20899 This tabulation is presented as an aid in the identification of the chemical structures of polycyclic aromatic hydrocarbons (PAHs). The Structure Index consists of two parts: (1) a cross index of named PAHs listed in alphabetical order, and (2) chemical structures including ring numbering, name(s), Chemical Abstract Service (CAS) Registry numbers, chemical formulas, molecular weights, and length-to-breadth ratios (L/B) and shape descriptors of PAHs listed in order of increasing molecular weight. Where possible, synonyms (including those employing alternate and/or obsolete naming conventions) have been included. Synonyms used in the Structure Index were compiled from a variety of sources including “Polynuclear Aromatic Hydrocarbons Nomenclature Guide,” by Loening, et al. [1], “Analytical Chemistry of Polycyclic Aromatic Compounds,” by Lee et al. [2], “Calculated Molecular Properties of Polycyclic Aromatic Hydrocarbons,” by Hites and Simonsick [3], “Handbook of Polycyclic Hydrocarbons,” by J. R. Dias [4], “The Ring Index,” by Patterson and Capell [5], “CAS 12th Collective Index,” [6] and “Aldrich Structure Index” [7]. In this publication the IUPAC preferred name is shown in large or bold type. -

ALABAMA SEAFOOD SURVEILLANCE SAMPLES NPH = Naphthalene, FLU = Fluorene, PHN = Phenanthrene, ANT = Anthracene, FLA = Fluoranthene
ALABAMA SEAFOOD SURVEILLANCE SAMPLES NPH = Naphthalene, FLU = Fluorene, PHN = Phenanthrene, ANT = Anthracene, FLA = Fluoranthene, Polycyclic Aromatic Hydrocarbon (PAH) and PYR = Pyrene, BaA = Benz(a)anthracene, CHR = Chrysene, BbF = Benzo(b)fluoranthene, DOSS Results Summary BkF = Benzo(k)fluoranthene, BaP = Benzo(a)pyrene, DBA = Dibenz(a,h)anthracene, IcdPy = Indeno(1,2,3-cd)pyrene, DOSS = Dioctylsulfosuccinate **The estimated maximum total PAH value represents a "worst case" estimate of the PAHs including alkyl homologs that could potentially be in the that happens to yield fluorescence responsesample. Results reported using FDACS Screening Method 521, based on It may include fluorescent compounds other than PAHs and background signal that happens to yield fluorescence response FDA LC Fluorescence Screening Method and FDACS DOSS Levels of Concern bases on FDA's Protocol for Interpretation and Use of Sensory Testing and Analytical Chemistry Results for Reopening In order to "PASS" Method 522 based on FDA's Determination of Levels of Concern (ppm) Oil-Impacted Areas closed to Seafood Harvesting. 7/26/10 samples must not Dioctylsulfosuccinate in Select Seafoods using LC/MS Shrimp and Crab 123 246 1846 246 185 1.32 1.32 13.2 0.132 0.132 1.32 61.5 500 exceed any Sorted by seafood type (crab, finfish, oyster, shrimp), Oysters 133 267 2000 267 200 1.43 143 1.43 14.3 0.143 0.143 1.43 66.5 500 FDA Levels of harvest area and sample # Finfish 32.7 65.3 490 65.3 49 0.35 35 0.35 0.35 0.035 0.035 0.35 16.35 100 Concern <LOD = less than Limit of Detection, -

Grignard Reaction: Synthesis of Triphenylmethanol
*NOTE: Grignard reactions are very moisture sensitive, so all the glassware in the reaction (excluding the work-up) should be dried in an oven with a temperature of > 100oC overnight. The following items require oven drying. They should be placed in a 150mL beaker, all labeled with a permanent marker. 1. 5mL conical vial (AKA: Distillation receiver). 2. Magnetic spin vane. 3. Claisen head. 4. Three Pasteur pipettes. 5. Two 1-dram vials (Caps EXCLUDED). 6. One 2-dram vial (Caps EXCLUDED). 7. Glass stirring rod 8. Adaptor (19/22.14/20) Grignard Reaction: Synthesis of Triphenylmethanol Pre-Lab: In the “equations” section, besides the main equations, also: 1) draw the equation for the production of the byproduct, Biphenyl. 2) what other byproduct might occur in the reaction? Why? In the “observation” section, draw data tables in the corresponding places, each with 2 columns -- one for “prediction” (by answering the following questions) and one for actual drops or observation. 1) How many drops of bromobenzene should you add? 2) How many drops of ether will you add to flask 2? 3) 100 µL is approximately how many drops? 4) What are the four signs of a chemical reaction? (Think back to Chem. 110) 5) How do the signs of a chemical reaction apply to this lab? The Grignard reaction is a useful synthetic procedure for forming new carbon- carbon bonds. This organometallic chemical reaction involves alkyl- or aryl-magnesium halides, known as Grignard 1 reagents. Grignard reagents are formed via the action of an alkyl or aryl halide on magnesium metal. -

Azulene—A Bright Core for Sensing and Imaging
molecules Review Azulene—A Bright Core for Sensing and Imaging Lloyd C. Murfin * and Simon E. Lewis Department of Chemistry, University of Bath, Bath BA2 7AY, UK; [email protected] * Correspondence: lloyd.murfi[email protected] Abstract: Azulene is a hydrocarbon isomer of naphthalene known for its unusual colour and fluores- cence properties. Through the harnessing of these properties, the literature has been enriched with a series of chemical sensors and dosimeters with distinct colorimetric and fluorescence responses. This review focuses specifically on the latter of these phenomena. The review is subdivided into two sec- tions. Section one discusses turn-on fluorescent sensors employing azulene, for which the literature is dominated by examples of the unusual phenomenon of azulene protonation-dependent fluorescence. Section two focuses on fluorescent azulenes that have been used in the context of biological sensing and imaging. To aid the reader, the azulene skeleton is highlighted in blue in each compound. Keywords: fluorescence; azulene; sensor; dosimeter; bioimaging; chemosensor; chemodosimeter 1. Introduction Azulene, 1, is an isomer of naphthalene, 2, composed of fused 5- and 7-membered ring systems (Figure1) and named for its vibrant blue colour. Unlike naphthalene, azulene is a non-alternant hydrocarbon, possessing nodal points at C-2 and C-6 of the HOMO and C-1 and C-3 of the LUMO [1]. The location of these nodes results in low electronic repulsion in the S1 singlet excited state, affording a relatively small HOMO-LUMO gap. Hence, the S0!S1 transition arises from absorption in the visible region. Conversely, in naphthalene, coefficient magnitudes remain consistent for each position in both the HOMO Citation: Murfin, L.C.; Lewis, S.E. -

Organic Chemistry Laboratory II Preparation of Triphenylmethanol (Grignard Reaction) Experiment Procedure (Printable Pdf Format)
grigdes http://www.organicchem.org/oc2web/lab/exp/grig/grigdesbenzophenone... Organic Chemistry II | Lecture | Laboratory Organic Chemistry Laboratory II Preparation of Triphenylmethanol (Grignard Reaction) Experiment Procedure (Printable pdf format) Introduction In this two-week experiment, triphenylmethanol will be synthesized through a Grignard reaction. Students will work individually to prepare the Grignard reagent by reacting bromobenzene with solid magnesium in diethylether. The prepared Grignard reagent, phenylmagnesium bromide, will then be combined with bezophenone to form the desired triphenylmethanol product. Procedure NOTE: All glassware must be extremely dry! Place glassware in the drying oven for at least 10 minutes before beginning the reaction. Preparation of the Grignard Reagent Add ~0.5g of magnesium turnings and a magnetic stir bar to a dry 100 ml round-bottomed flask, and place the flask in the drying oven for 30 minutes. Remove the flask from the oven, clamp it to a ring stand, insert a reflux condenser, fit the flask into a heating mantle and immediately attach a drying tube to the top of the reflux condenser. Position the flask in the heating mantle on a stirrer/hotplate. Set the reaction up on the side of the hood near to the faucet labeled for distillation. Allow the reaction flask to cool to room temperature completely before proceeding. While the flask is cooling, place a 150ml beaker into the drying oven. While the flask is cooling, add 3.5 g of dry bromobenzene to a dry 50 mL Erlenmeyer flask and dissolve it in 5.0 mL of anhydrous diethyl ether. Use a glass powder funnel to dispense the bromobenze and ether to the Erlenmyer flask. -
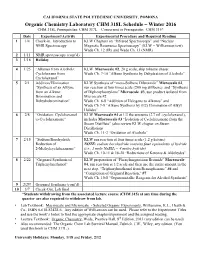
318 Lab Schedule
CALIFORNIA STATE POLYTECHNIC UNIVERSITY, POMONA Organic Chemistry Laboratory CHM 318L Schedule – Winter 2016 CHM 318L Prerequisites: CHM 317L Concurrent or Prerequisite: CHM 315* Date Experiment/Activity Experimental Procedure and Required Reading 1 1/4 Check-in. Introduction to KLW Chapters on “Infrared Spectroscopy” and “Nuclear NMR Spectroscopy Magnetic Resonance Spectroscopy” (KLW = Williamson text) Wade Ch. 12 (IR) and Wade Ch. 13 (NMR) 2 1/11 NMR spectroscopy (cont’d) 3 1/18 Holiday 4 1/25 “Alkenes from Alcohols: KLW Macroscale #2, 20 g scale, skip toluene chaser Cyclohexene from Wade Ch. 7-10 “Alkene Synthesis by Dehydration of Alcohols” Cyclohexanol” 5 2/1 Addition/Elimination KLW Synthesis of “meso-Stilbene Dibromide” Microscale #2, “Synthesis of an Alkyne run reaction at four times scale (200 mg stillbene); and “Synthesis from an Alkene: of Diphenylacetylene” Microscale #3, use product isolated from Bromination and Microscale #2 Dehydrobromination” Wade Ch. 8-8 “Addition of Halogens to Alkenes” and Wade Ch 7-9 “Alkene Synthesis by (E2) Elimination of Alkyl Halides” 6 2/8 “Oxidation: Cyclohexanol KLW Macroscale #4 at 1/3 the amounts (2.7 mL cyclohexanol), to Cyclohexanone” includes Macroscale #3 “Isolation of Cyclohexanone from the Steam Distillate” (also review KLW chapter on Steam Distillation) Wade Ch. 11-2 “Oxidation of Alcohols” 7 2/15 “Sodium Borohydride KLW run reaction at four times scale (1.2 g ketone) Reduction of NOTE: sodium borohydride contains four equivalents of hydride 2-Methylcyclohexanone” (i.e., 1 mole NaBH4 = 4 moles hydride) Wade Ch. 10-11 & 18-20 “Reductions of Ketones & Aldehydes” 8 2/22 “Grignard Synthesis of KLW preparation of “Phenylmagnesium Bromide” Macroscale Triphenylmethanol” #4, run reaction at 1/2 scale and then use the entire amount in the next step, “Triphenylmethanol from Benzophenone” #6 and “Completion of Grignard Reaction,” #7 Wade Ch. -

Revised Group Additivity Values for Enthalpies of Formation (At 298 K) of Carbon– Hydrogen and Carbon–Hydrogen–Oxygen Compounds
Revised Group Additivity Values for Enthalpies of Formation (at 298 K) of Carbon– Hydrogen and Carbon–Hydrogen–Oxygen Compounds Cite as: Journal of Physical and Chemical Reference Data 25, 1411 (1996); https://doi.org/10.1063/1.555988 Submitted: 17 January 1996 . Published Online: 15 October 2009 N. Cohen ARTICLES YOU MAY BE INTERESTED IN Additivity Rules for the Estimation of Molecular Properties. Thermodynamic Properties The Journal of Chemical Physics 29, 546 (1958); https://doi.org/10.1063/1.1744539 Critical Evaluation of Thermochemical Properties of C1–C4 Species: Updated Group- Contributions to Estimate Thermochemical Properties Journal of Physical and Chemical Reference Data 44, 013101 (2015); https:// doi.org/10.1063/1.4902535 Estimation of the Thermodynamic Properties of Hydrocarbons at 298.15 K Journal of Physical and Chemical Reference Data 17, 1637 (1988); https:// doi.org/10.1063/1.555814 Journal of Physical and Chemical Reference Data 25, 1411 (1996); https://doi.org/10.1063/1.555988 25, 1411 © 1996 American Institute of Physics for the National Institute of Standards and Technology. Revised Group Additivity Values for Enthalpies of Formation (at 298 K) of Carbon-Hydrogen and Carbon-Hydrogen-Oxygen Compounds N. Cohen Thermochemical Kinetics Research, 6507 SE 31st Avenue, Portland, Oregon 97202-8627 Received January 17, 1996; revised manuscript received September 4, 1996 A program has been undertaken for the evaluation and revision of group additivity values (GAVs) necessary for predicting, by means of Benson's group additivity method, thermochemical properties of organic molecules. This review reports on the portion of that program dealing with GAVs for enthalpies of formation at 298.15 K (hereinafter abbreviated as 298 K) for carbon-hydrogen and carbon-hydrogen-oxygen compounds. -

Polycyclic Aromatic Hydrocarbons (Pahs)
Polycyclic Aromatic Hydrocarbons (PAHs) Factsheet 4th edition Donata Lerda JRC 66955 - 2011 The mission of the JRC-IRMM is to promote a common and reliable European measurement system in support of EU policies. European Commission Joint Research Centre Institute for Reference Materials and Measurements Contact information Address: Retiewseweg 111, 2440 Geel, Belgium E-mail: [email protected] Tel.: +32 (0)14 571 826 Fax: +32 (0)14 571 783 http://irmm.jrc.ec.europa.eu/ http://www.jrc.ec.europa.eu/ Legal Notice Neither the European Commission nor any person acting on behalf of the Commission is responsible for the use which might be made of this publication. Europe Direct is a service to help you find answers to your questions about the European Union Freephone number (*): 00 800 6 7 8 9 10 11 (*) Certain mobile telephone operators do not allow access to 00 800 numbers or these calls may be billed. A great deal of additional information on the European Union is available on the Internet. It can be accessed through the Europa server http://europa.eu/ JRC 66955 © European Union, 2011 Reproduction is authorised provided the source is acknowledged Printed in Belgium Table of contents Chemical structure of PAHs................................................................................................................................. 1 PAHs included in EU legislation.......................................................................................................................... 6 Toxicity of PAHs included in EPA and EU -

NON-TARGET ANALYSIS of BIOREMEDIATED SOIL Zhenyu
NON-TARGET ANALYSIS OF BIOREMEDIATED SOIL Zhenyu Tian A dissertation submitted to the faculty at the University of North Carolina at Chapel Hill in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Department of Environmental Sciences and Engineering in the Gillings School of Global Public Health. Chapel Hill 2018 Approved by: Michael D. Aitken Wanda M. Bodnar Avram Gold Kun Lu Jason D. Surratt © 2018 Zhenyu Tian ALL RIGHTS RESERVED ii ABSTRACT Zhenyu Tian: Non-target Analysis of Bioremediated Soil (Under the direction of Michael D. Aitken) Polycyclic aromatic hydrocarbons (PAHs) are ubiquitous pollutants of environmental concern. Bioremediation, relying on stimulation of natural microbial degradation processes, is a well-established technology to clean up PAH-contaminated soils. However, bioremediation does not necessarily lead to a reduction in soil toxicity. PAH-contaminated sites are affected by extremely complex mixtures, like coal tar or creosote, and biotransformation products or co- occurring compounds can also contribute to the overall toxicological effects of contaminated soil before and after bioremediation. Therefore, the objective of this dissertation was to use non- target analysis workflows to identify the genotoxic transformation products, important co- occurring pollutants, and the unrecognized biotransformation pathways that could contribute to explain the toxicological effects observed beyond parent PAHs. To identify the source(s) of increased genotoxicity in bioremediated soil, we pursued a non-target analytical approach combining effect-directed analysis (EDA) and metabolite profiling to compare extracts of PAH-contaminated soil before and after bioremediation. A compound with the composition C15H8O2 and four methylated homologues were shown to accumulate as a result of bioreactor treatment, and the C15H8O2 compound was determined to be genotoxic. -

Oxidation of 9-Fluorenol to 9-Fluorenone with Sodium Hypochlorite
CHEM 2229 EXP 1: Oxidation of 9-Fluorenol to 9-Fluorenone with Sodium Hypochlorite Objective: In this experiment you will learn how to perform an oxidation reaction by oxidizing an alcohol (9- hydroxyfluorene) to a ketone (9-fluorenone) using sodium hypochlorite in an acidic environment; how to perform TLC to monitor a reaction; how to perform an extraction to isolate a product; and, how to verify purity of a product using TLC and melting point. * Chromatography is a useful method for separating components of a mixture of compounds based on their polarity. Thin layer chromatography is especially useful for determining the number of components in a mixture, the identity of the compounds, and the purity of a compound. **Solvent Extraction is also known as Liquid–liquid extraction (LLE) or partitioning. It is a method used to separate compounds based on their relative solubilities in two different immiscible liquids: usually the polar solvent water and a non-polar organic solvent. Immiscible means that the liquids do not mix and because of this form two distinct layers. ***The melting point (MP) is a physical property of a solid also used for the purpose of identification and purity determination. Reading Assignment: OCLT: OCLT, pp. 83-108 (chromatography generalities & TLC), pp. 368-369 (TLC technique summary), pp. 203-246 (extraction); pp. 376-381 (extraction illustrations); 366 (vacuum filtration); and 309-315 (melting point). Solomons Organic Chemistry, 12th ed. (Note: Pages correspond to 12th ed.) pp. 542-547 (12.4 Oxidation of Alcohols) Concepts: Acids, Bases, Decantation, Drying Agents, Exothermic Reactions, Extraction, Half Cell Method, Oxidation/Reduction, Oxidizing Agents, Reducing Agents, Reflux, Salting Out Chemicals: acetic acid (glacial), acetone, 9-fluorenol, 9-fluorenone, hexane, sodium bicarbonate, sodium chloride, sodium hypochlorite (aq soln) / bleach, sodium sulfate Safety Precautions: Wear chemical splash-proof goggles and appropriate attire at all times.