Collapse of the Hepatic Gene Regulatory Network in the Absence of Foxa Factors
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Serum Concentrations of Afamin Are Elevated in Patients with Polycystic Ovary Syndrome
AKo¨ ninger et al. Afamin in patients with PCOS 1–7 3:120 Research Open Access Serum concentrations of afamin are elevated in patients with polycystic ovary syndrome Angela Ko¨ ninger, Philippos Edimiris, Laura Koch, Antje Enekwe, Claudia Lamina1, Sabine Kasimir-Bauer, Rainer Kimmig and Hans Dieplinger1,2 Correspondence Department of Gynecology and Obstetrics, University of Duisburg-Essen, D-45122 Essen, Germany should be addressed 1Division of Genetic Epidemiology, Department of Medical Genetics, Molecular and Clinical Pharmacology, to H Dieplinger Innsbruck Medical University, Scho¨ pfstrasse 41, A-6020 Innsbruck, Austria Email 2Vitateq Biotechnology GmbH, A-6020 Innsbruck, Austria [email protected] Abstract Oxidative stress seems to be present in patients with polycystic ovary syndrome (PCOS). Key Words The aim of this study was to evaluate the correlation between characteristics of PCOS and " vitamin E-binding protein serum concentrations of afamin, a novel binding protein for the antioxidant vitamin E. " afamin A total of 85 patients with PCOS and 76 control subjects were investigated in a pilot cross- " polycystic ovary syndrome sectional study design between 2009 and 2013 in the University Hospital of Essen, Germany. " oxidative stress Patients with PCOS were diagnosed according to the Rotterdam ESHRE/ASRM-sponsored " insulin resistance PCOS Consensus Workshop Group. Afamin and diagnostic parameters of PCOS were determined at early follicular phase. Afamin concentrations were significantly higher in Endocrine Connections patients with PCOS than in controls (odds ratio (OR) for a 10 mg/ml increase in afaminZ1.3, 95% CIZ1.08–1.58). This difference vanished in a model adjusting for age, BMI, free testosterone index (FTI), and sex hormone-binding globulin (SHBG) (ORZ1.05, 95% CIZ0.80–1.38). -

Antisense Afp Transcripts in Mouse Liver and Their Potential Role in Afp Gene Regulation
University of Kentucky UKnowledge Theses and Dissertations--Microbiology, Microbiology, Immunology, and Molecular Immunology, and Molecular Genetics Genetics 2017 ANTISENSE AFP TRANSCRIPTS IN MOUSE LIVER AND THEIR POTENTIAL ROLE IN AFP GENE REGULATION Maria S. Dixon University of Kentucky, [email protected] Digital Object Identifier: https://doi.org/10.13023/ETD.2017.356 Right click to open a feedback form in a new tab to let us know how this document benefits ou.y Recommended Citation Dixon, Maria S., "ANTISENSE AFP TRANSCRIPTS IN MOUSE LIVER AND THEIR POTENTIAL ROLE IN AFP GENE REGULATION" (2017). Theses and Dissertations--Microbiology, Immunology, and Molecular Genetics. 14. https://uknowledge.uky.edu/microbio_etds/14 This Doctoral Dissertation is brought to you for free and open access by the Microbiology, Immunology, and Molecular Genetics at UKnowledge. It has been accepted for inclusion in Theses and Dissertations--Microbiology, Immunology, and Molecular Genetics by an authorized administrator of UKnowledge. For more information, please contact [email protected]. STUDENT AGREEMENT: I represent that my thesis or dissertation and abstract are my original work. Proper attribution has been given to all outside sources. I understand that I am solely responsible for obtaining any needed copyright permissions. I have obtained needed written permission statement(s) from the owner(s) of each third-party copyrighted matter to be included in my work, allowing electronic distribution (if such use is not permitted by the fair use doctrine) which will be submitted to UKnowledge as Additional File. I hereby grant to The University of Kentucky and its agents the irrevocable, non-exclusive, and royalty-free license to archive and make accessible my work in whole or in part in all forms of media, now or hereafter known. -
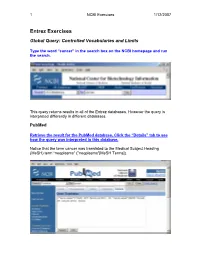
Entrez Exercises
1 NCBI Exercises 1/12/2007 Entrez Exercises Global Query: Controlled Vocabularies and Limits Type the word “cancer” in the search box on the NCBI homepage and run the search. This query returns results in all of the Entrez databases. However the query is interpreted differently in different databases. PubMed Retrieve the result for the PubMed database. Click the “Details” tab to see how the query was interpreted in this database. Notice that the term cancer was translated to the Medical Subject Heading (MeSH) term “neoplasms” ("neoplasms"[MeSH Terms]). 2 NCBI Exercises 1/12/2007 MeSH is a controlled vocabulary that is used to index all articles in PubMed. In the details box, edit the query to remove the portion that searched for cancer as a text word and run the search. Notice that the number of articles retrieved has changed. These will be a more relevant set of results. You can force the PubMed engine to only search the MeSH vocabulary or specify any other indexed field through the “Limits” tab. Use the Web browser’s back button to return to the Global query page and retrieve the PubMed results again. Now click on the “Limits” tab. Select “MeSH terms” from the first drop-down menu, the one headed by “All Fields”. Now run the search with the limit in place and check the “Details” tab to verify that only the MeSH term translation was used. Nucleotide Use the Web browser’s back button to return to the Global query page. Retrieve the results for the nucleotide database. 3 NCBI Exercises 1/12/2007 The Nucleotide database in the Global query is now three separate databases. -

Page 1 of 76 Diabetes Diabetes Publish Ahead of Print, Published Online September 14, 2020
Page 1 of 76 Diabetes Peters, Annette; Helmholtz Center Munich German Research Center for Environmental Health, Epidemiology Institute Waldenberger, Melanie; Helmholtz Center Munich German Research Center for Environmental Health, Molecular Epidemiology Diabetes Publish Ahead of Print, published online September 14, 2020 Diabetes Page 2 of 76 Deciphering the Plasma Proteome of Type 2 Diabetes Mohamed A. Elhadad1,2,3 MSc., Christian Jonasson4,5 PhD, Cornelia Huth2,6 PhD, Rory Wilson1,2 MSc, Christian Gieger1,2,6 PhD, Pamela Matias1,2,3 MSc, Harald Grallert1,2,6 PhD, Johannes Graumann7,8 PhD, Valerie Gailus-Durner9 PhD, Wolfgang Rathmann6,10 MD, Christine von Toerne11 PhD, Stefanie M. Hauck11 PhD, Wolfgang Koenig3,12,13 MD, FRCP, FESC, FACC, FAHA, Moritz F. Sinner3,14 MD, MPH, Tudor I Oprea15,16,17 MD, PhD, Karsten Suhre18 PhD, Barbara Thorand2,6 PhD, Kristian Hveem4,5 PhD, Annette Peters2,3,6,19 PhD, Melanie Waldenberger1,2,3 PhD 1. Research Unit of Molecular Epidemiology, Helmholtz Zentrum München, German Research Center for Environmental Health, Neuherberg, Germany. 2. Institute of Epidemiology, Helmholtz Zentrum München, German Research Center for Environmental Health, Neuherberg, Germany 3. German Research Center for Cardiovascular Disease (DZHK), Partner site Munich Heart Alliance, Germany 4. K.G. Jebsen Center for Genetic Epidemiology, Department of Public Health, NTNU - Norwegian University of Science and Technology, Trondheim, Norway 5. HUNT Research Center, Department of Public Health, NTNU - Norwegian University of Science and Technology, Levanger, Norway 6. German Center for Diabetes Research (DZD), München-Neuherberg, Ingolstädter Landstr. 1, 85764, Neuherberg, Germany 7. Biomolecular Mass Spectrometry, Max Planck Institute for Heart and Lung Research, Ludwigstrasse 43, Bad Nauheim 61231, Germany 8. -

Human Afamin Elisa
HUMAN AFAMIN ELISA Product Data Sheet Cat. No.: RD194428100R For Research Use Only Page 1 of 24 ENG.002.A CONTENTS 1. INTENDED USE 3 2. STORAGE, EXPIRATION 3 3. INTRODUCTION 4 4. TEST PRINCIPLE 4 5. PRECAUTIONS 5 6. TECHNICAL HINTS 5 7. REAGENT SUPPLIED 6 8. MATERIAL REQUIRED BUT NOT SUPPLIED 6 9. PREPARATION OF REAGENTS 7 10. PREPARATION OF SAMPLES 8 11. ASSAY PROCEDURE 9 12. CALCULATIONS 11 13. PERFORMANCE CHARACTERISTICS 12 14. DEFINITION OF THE STANDARD 15 15. PRELIMINARY POPULATION DATA 15 16. METHOD COMPARISON 16 17. TROUBLESHOOTING AND FAQS 18 18. REFERENCES 19 19. EXPLANATION OF SYMBOLS 20 This kit is manufactured by: BioVendor – Laboratorní medicína a.s. under the contract with Vitateq Biotechnology GmbH Use only the current version of Product Data Sheet enclosed with the kit! Page 2 of 24 ENG.002.A 1. INTENDED USE The RD194428100R Human Afamin ELISA is a sandwich enzyme immunoassay for the quantitative measurement of human afamin. Features • It is intended for research use only. • The total assay time is less than 3 hours. • The kit measures afamin in serum and plasma (EDTA, citrate, heparin). • Assay format is 96 wells. • Standard is a serum-based protein. • Components of the kit are provided ready to use, concentrated or lyophilized. 2. STORAGE, EXPIRATION Store the complete kit at 2–8°C. Under these conditions, the kit is stable until the expiration date (see label on the box). For stability of opened reagents see Chapter 9. Page 3 of 24 ENG.002.A 3. INTRODUCTION Mature human afamin, the product of the AFM gene, is a single chain 75 kDa protein consisting of 578 amino acid residues. -
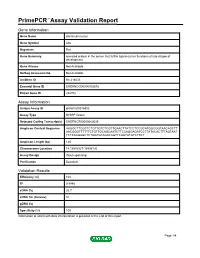
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name afamin precursor Gene Symbol Afm Organism Rat Gene Summary secreted protein in the serum that fulfills ligand-carrier functions at late stages of development Gene Aliases Not Available RefSeq Accession No. Not Available UniGene ID Rn.214035 Ensembl Gene ID ENSRNOG00000002878 Entrez Gene ID 282708 Assay Information Unique Assay ID qRnoCID0016852 Assay Type SYBR® Green Detected Coding Transcript(s) ENSRNOT00000060535 Amplicon Context Sequence AAGGCTTCGCTCTGTTGTCTCGTTGAACTTATCCTCCGCATGGCGGTAACAGCTT AACGGGTTTTTTCTGTTGCAGCAATCTTCCAGGAGATCCTTATACACTTTAGTAAT TCTTAGGAGCTCTGGTGTAGACAGTTCAGTATGTCTTCT Amplicon Length (bp) 120 Chromosome Location 14:18993927-18996130 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 103 R2 0.9998 cDNA Cq 32.7 cDNA Tm (Celsius) 81 gDNA Cq Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 1/4 PrimePCR™Assay Validation Report Afm, Rat Amplification Plot Amplification of cDNA generated from 25 ng of universal reference RNA Melt Peak Melt curve analysis of above amplification Standard Curve Standard curve generated using 20 million copies of template diluted 10-fold to 20 copies Page 2/4 PrimePCR™Assay Validation Report Products used to generate validation data Real-Time PCR Instrument CFX384 Real-Time PCR Detection System Reverse Transcription Reagent iScript™ Advanced cDNA Synthesis Kit for RT-qPCR Real-Time PCR Supermix SsoAdvanced™ SYBR® Green Supermix Experimental Sample qPCR Reference Total RNA Data Interpretation Unique Assay ID This is a unique identifier that can be used to identify the assay in the literature and online. Detected Coding Transcript(s) This is a list of the Ensembl transcript ID(s) that this assay will detect. Details for each transcript can be found on the Ensembl website at www.ensembl.org. -

Genome-Wide Analysis of Pax8 Binding Provides New Insights Into Thyroid Functions
Ruiz-Llorente et al. BMC Genomics 2012, 13:147 http://www.biomedcentral.com/1471-2164/13/147 RESEARCH ARTICLE Open Access Genome-wide analysis of Pax8 binding provides new insights into thyroid functions Sergio Ruiz-Llorente1,2, Enrique Carrillo SantadePau1,3,4, Ana Sastre-Perona1, Cristina Montero-Conde1,2, Gonzalo Gómez-López3, James A Fagin2, Alfonso Valencia3, David G Pisano3 and Pilar Santisteban1* Abstract Background: The transcription factor Pax8 is essential for the differentiation of thyroid cells. However, there are few data on genes transcriptionally regulated by Pax8 other than thyroid-related genes. To better understand the role of Pax8 in the biology of thyroid cells, we obtained transcriptional profiles of Pax8-silenced PCCl3 thyroid cells using whole genome expression arrays and integrated these signals with global cis-regulatory sequencing studies performed by ChIP-Seq analysis Results: Exhaustive analysis of Pax8 immunoprecipitated peaks demonstrated preferential binding to intragenic regions and CpG-enriched islands, which suggests a role of Pax8 in transcriptional regulation of orphan CpG regions. In addition, ChIP-Seq allowed us to identify Pax8 partners, including proteins involved in tertiary DNA structure (CTCF) and chromatin remodeling (Sp1), and these direct transcriptional interactions were confirmed in vivo. Moreover, both factors modulate Pax8-dependent transcriptional activation of the sodium iodide symporter (Nis) gene promoter. We ultimately combined putative and novel Pax8 binding sites with actual target gene expression regulation to define Pax8-dependent genes. Functional classification suggests that Pax8-regulated genes may be directly involved in important processes of thyroid cell function such as cell proliferation and differentiation, apoptosis, cell polarity, motion and adhesion, and a plethora of DNA/protein-related processes. -

Mechanisms of Sodium–Glucose Cotransporter 2 Inhibition
Diabetes Care Volume 43, September 2020 2183 – Ele Ferrannini,1 Ashwin C. Murthy,2 Mechanisms of Sodium Glucose Yong-hoLee,3 ElzaMuscelli,1 SophieWeiss,4 Rachel M. Ostroff,4 Naveed Sattar,5 Cotransporter 2 Inhibition: Stephen A. Williams,4 and Peter Ganz6 Insights From Large-Scale Proteomics Diabetes Care 2020;43:2183–2189 | https://doi.org/10.2337/dc20-0456 OBJECTIVE To assess the effects of empagliflozin, a selective sodium–glucose cotransporter 2 (SGLT2) inhibitor, on broad biological systems through proteomics. RESEARCH DESIGN AND METHODS Aptamer-based proteomics was used to quantify 3,713 proteins in 144 paired plasma samples obtained from 72 participants across the spectrum of glucose tolerance before and after 4 weeks of empagliflozin 25 mg/day. The biology of the plasma proteins significantly changed by empagliflozin (at false discovery rate– corrected P < 0.05) was discerned through Ingenuity Pathway Analysis. RESULTS Empagliflozin significantly affected levels of 43 proteins, 6 related to cardiomyocyte function(fattyacid–bindingprotein3and4[FABPA],neurotrophic receptortyrosine kinase, renin, thrombospondin 4, and leptin receptor), 5 to iron handling (ferritin heavy chain 1, transferrin receptor protein 1, neogenin, growth differentiation CARDIOVASCULAR AND METABOLIC RISK factor 2 [GDF2], and b2-microglobulin), and 1 to sphingosine/ceramide metabolism 1CNR Institute of Clinical Physiology, Pisa, Italy (neutral ceramidase), a known pathway of cardiovascular disease. Among the 2 fi Cardiovascular Division, Department of Medi- protein changes achieving the strongest statistical signi cance, insulin-like binding cine, Hospital of the University of Pennsylvania, factor protein-1 (IGFBP-1), transgelin-2, FABPA, GDF15, and sulphydryl oxidase Philadelphia, PA 2 precursor were increased, while ferritin, thrombospondin 3, and Rearranged 3Department of Medicine, Severance Hospital, during Transfection (RET) were decreased by empagliflozin administration. -

Afamin Antibody / AFM (RQ5792)
Afamin Antibody / AFM (RQ5792) Catalog No. Formulation Size RQ5792 0.5mg/ml if reconstituted with 0.2ml sterile DI water 100 ug Bulk quote request Availability 1-3 business days Species Reactivity Human Format Antigen affinity purified Clonality Polyclonal (rabbit origin) Isotype Rabbit IgG Purity Affinity purified Buffer Lyophilized from 1X PBS with 2% Trehalose and 0.025% sodium azide UniProt P43652 Applications Western blot : 0.5-1ug/ml Immunohistochemistry : 1-2ug/ml Flow cytometry : 1-3ug/million cells Direct ELISA : 0.1-0.5ug/ml Limitations This AFM antibody is available for research use only. IHC staining of FFPE human ovarian cancer with Afamin antibody. HIER: boil tissue sections in pH8 EDTA for 20 min and allow to cool before testing. IHC staining of FFPE human liver cancer with Afamin antibody. HIER: boil tissue sections in pH8 EDTA for 20 min and allow to cool before testing. Western blot testing of human plasma lysate with Afamin antibody. Predicted molecular weight: ~69/87 kDa (unmodified/glycosylated). Flow cytometry testing of human HL-60 cells with Afamin antibody at 1ug/million cells (blocked with goat sera); Red=cells alone, Green=isotype control, Blue= Afamin antibody. Description Afamin is a protein that in humans is encoded by the AFM gene. This gene is a member of the albumin gene family, which is comprised of four genes that localize to chromosome 4 in a tandem arrangement. These four genes encode structurally-related serum transport proteins that are known to be evolutionarily related. The protein encoded by this gene is regulated developmentally, expressed in the liver and secreted into the bloodstream. -

Organoid Profiling Identifies Common Responders to Chemotherapy in Pancreatic Cancer
Published OnlineFirst May 31, 2018; DOI: 10.1158/2159-8290.CD-18-0349 RESEARCH ARTICLE Organoid Profi ling Identifi es Common Responders to Chemotherapy in Pancreatic Cancer Hervé Tiriac 1 , Pascal Belleau 1 , Dannielle D. Engle 1 , Dennis Plenker1 , Astrid Deschênes 1 , Tim D. D. Somerville1 , Fieke E. M. Froeling 1 , Richard A. Burkhart 2 , Robert E. Denroche 3 , Gun-Ho Jang 3 , Koji Miyabayashi 1 , C. Megan Young1 , 4 , Hardik Patel 1 , Michelle Ma 1 , Joseph F. LaComb 5 , Randze Lerie D. Palmaira 6 , Ammar A. Javed2 , Jasmine C. Huynh 7 , Molly Johnson 8 , Kanika Arora 8 , Nicolas Robine 8 , Minita Shah 8 , Rashesh Sanghvi8 , Austin B. Goetz 9 , Cinthya Y. Lowder 9 , Laura Martello10 , Else Driehuis 11 , 12 , Nicolas LeComte6 , Gokce Askan 6 , Christine A. Iacobuzio-Donahue 6 , Hans Clevers 11 , 12 , 13 , Laura D. Wood 14 , Ralph H. Hruban 14 , Elizabeth Thompson14 , Andrew J. Aguirre 15 , Brian M. Wolpin 15 , Aaron Sasson 16 , Joseph Kim 16 , Maoxin Wu 17 , Juan Carlos Bucobo5 , Peter Allen6 , Divyesh V. Sejpal 18 , William Nealon 19 , James D. Sullivan 19 , Jordan M. Winter 9 , Phyllis A. Gimotty 20 , Jean L. Grem 21 , Dominick J. DiMaio 22 , Jonathan M. Buscaglia 5 , Paul M. Grandgenett 23 , Jonathan R. Brody9 , Michael A. Hollingsworth 23 , Grainne M. O’Kane 24 , Faiyaz Notta 3 , Edward Kim 7 , James M. Crawford25 , Craig Devoe 26 , Allyson Ocean 27 , Christopher L. Wolfgang 2 , Kenneth H. Yu 6 , Ellen Li 5 , Christopher R. Vakoc1 , Benjamin Hubert 8 , Sandra E. Fischer 28 , 29 , Julie M. Wilson 3 , Richard Moffi tt 16 , 30 , Jennifer Knox24 , Alexander Krasnitz 1 , Steven Gallinger 3 , 24 , 31 , 32 , and David A. -

Supplemental Table 1 Proteins Only Present in VLDL Fraction Entrez Gene Name Coagulation System Extrinsic Prothrombin Activation
Supplemental Table 1 Proteins only present in VLDL fraction Intrinsic Extrinsic Prothrombi Entrez Gene Name Coagulation Prothrombin n Cellular System Activation Activation location 1 ADP-ribosylation factor interacting protein 1 - - - Cytoplasm 2 afamin - - - Extracellular 3 alpha-1-B glycoprotein - - - Extracellular 4 alpha-2-macroglobulin + - - Extracellular 5 alpha-2-macroglobulin-like 1 - - - Cytoplasm 6 angiotensinogen (serpin peptidase inhibitor, clade A, member 8) - - - Extracellular 7 angiotensinogen (serpin peptidase inhibitor, clade A, member 8) - - - Extracellular 8 apolipoprotein B - - - Extracellular 9 apolipoprotein B - - - Extracellular 10 apolipoprotein E - - - Extracellular 11 apolipoprotein E - - - Extracellular 12 apolipoprotein E - - - Extracellular 13 ArfGAP with GTPase domain, ankyrin repeat and PH domain 6 - - - Other 14 ATP-binding cassette, sub-family A (ABC1), member 1 - - - Plasma Membrane 15 ATP-binding cassette, sub-family C (CFTR/MRP), member 12 - - - Cytoplasm 16 attractin - - - Extracellular 17 Bardet-Biedl syndrome 4 - - - Cytoplasm 18 carboxypeptidase N, polypeptide 2 - - - Extracellular 19 caspase 8, apoptosis-related cysteine peptidase - - - Nucleus 20 Cbl proto-oncogene, E3 ubiquitin protein ligase - - - Nucleus 21 ceruloplasmin (ferroxidase) - - - Extracellular 22 chemokine (C-X-C motif) ligand 2 - - - Extracellular 23 cholinergic receptor, muscarinic 2 - - - Plasma 1 Membrane 24 chromosome 6 open reading frame 163 - - - Other 25 clathrin, heavy chain (Hc) - - - Plasma Membrane 26 coagulation factor -
Study of Urine Extracellular Vesicles-Derived Protein Biomarkers for the Non-Invasive Monitoring of Kidney-Transplanted Patients
ADVERTIMENT. Lʼaccés als continguts dʼaquesta tesi queda condicionat a lʼacceptació de les condicions dʼús establertes per la següent llicència Creative Commons: http://cat.creativecommons.org/?page_id=184 ADVERTENCIA. El acceso a los contenidos de esta tesis queda condicionado a la aceptación de las condiciones de uso establecidas por la siguiente licencia Creative Commons: http://es.creativecommons.org/blog/licencias/ WARNING. The access to the contents of this doctoral thesis it is limited to the acceptance of the use conditions set by the following Creative Commons license: https://creativecommons.org/licenses/?lang=en Study of urine extracellular vesicles-derived protein biomarkers for the non-invasive monitoring of kidney- transplanted patients Laura Carreras Planella Doctoral thesis Badalona, 20th October 2020 Thesis directors: Francesc Enric Borràs Serres, PhD Maria Isabel Troya Saborido, PhD 2 PhD programme in Advanced Immunology Department of Cellular Biology, Physiology and Immunology Universitat Autònoma de Barcelona Study of urine extracellular vesicles-derived protein biomarkers for the non-invasive monitoring of kidney-transplanted patients Estudi de biomarcadors proteics derivats de vesícules extracel·lulars de la orina per la monitorització no invasiva de pacients trasplantats de ronyó Thesis presented by Laura Carreras Planella to qualify for the PhD degree in Advanced Immunology by the Universitat Autònoma de Barcelona The presented work has been performed in the ReMAR-IVECAT group at the Germans Trias i Pujol Health Sciences Research Institute (IGTP) under the supervision of Dr. Francesc Enric Borràs Serres, as tutor and co-director, and Dr. Maria Isabel Troya Saborido as co-director. 3 Laura Carreras was sponsored by the Spanish Government with an FPU grant (“Formación de Personal Universitario”, FPU17/01444) and by La Fundació Cellex during the development of the PhD project.