Cyclin D1 and CDK4 Activity Contribute to the Undifferentiated Phenotype in Neuroblastoma
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Microarray Analysis of Gene Expression in the Ovarian Cancer Cell Line HO-8910 with Silencing of the ZNF217 Gene
MOLECULAR MEDICINE REPORTS 2: 851-855, 2009 851 Microarray analysis of gene expression in the ovarian cancer cell line HO-8910 with silencing of the ZNF217 gene GUIQIN SUN1, JINGXIA QIN1, YUWEN QIU1, YUNFEI GAO1, YANHONG YU1, QINKAI DENG2 and MEI ZHONG1 1Department of Obstetrics and Gynecology, Nanfang Hospital, Southern Medical University; 2Department of Bioinformatics, Southern Medical University, Guangzhou 510515, Guangdong, P.R. China Received April 30, 2009; Accepted July 7, 2009 DOI: 10.3892/mmr_00000183 Abstract. Zinc-finger protein 217 (ZNF217), which is over- late stages (stage III or IV) and have a 5-year survival rate of expressed during cancer progression, can promote tumor cell less than 30% (1). Ovarian cancer is a heterogeneous disease immortalization. To examine the function of ZNF217, a global with respect to its histopathology, molecular biology and expression profile was carried out using Affymetrix Gene clinical outcome. Though studies on the etiology of ovarian Chip analysis with HG-U133 plus 2.0 arrays in the ovarian cancer and susceptibility to the disease have shown that the cancer cell line HO-8910 after silencing of the ZNF217 gene. mechanisms of carcinogenesis and cancer development are The results were analyzed using the Gene Ontology program associated with genetic, epigenetic and environmental factors, to investigate the functional network affected by ZNF217 the pathogenesis of ovarian cancer remains unclear (2). in ovarian cancer cells. Changes in the mRNA expression Among the current methods for the identification of gene of the affected genes were confirmed by real-time reverse expression profiles in cancer, microarray analysis is the most transcriptase-polymerase chain reaction (RT-PCR). -

In Silico Analysis Identifies Genes Common Between Five Primary Gastrointestinal Cancer Sites with Potential Clinical Applications
ORIGINAL ARTICLE Annals of Gastroenterology (2014) 27, 1-14 In silico analysis identifies genes common between five primary gastrointestinal cancer sites with potential clinical applications Subhankar Chakraborty University of Nebraska Medical Center, Omaha, NE, USA Abstract Background Previous studies have investigated differential gene expression in gastrointes- tinal (GI) epithelial cancers by microarray. The aim of the present study was to use data from the Oncomine database to identify genes that share a similar differential expression in two or more primary GI cancer sites. Methods Five thousand of the most differentially expressed genes in epithelial cancers (com- pared to normal tissue) arising in the pancreas, liver, stomach, esophagus or colorectum were identified (1,000 per primary site) from Oncomine. Using Venn diagrams, genes common to two or more primary GI sites were identified. Functional and pathway analysis was performed on genes that were similarly expressed in ≥3 of the five areas of the GI tract. Results Forty six studies comprising 5,876 samples were included. Overall, 90.6% genes were unique to the respective primary sites, 7.4% shared between two GI primary sites, 1.8% between three and 0.2% between four GI primary sites. Pancreatic and hepatocellular cancers (HCC) shared most number of upregulated genes (N=66) while HCC and gastric cancer shared most downregulated genes (N=59). Genes encoding enzymes comprised the most commonly shared genes between GI primary sites (30.4% of upregulated and 63.2% of downregulated genes). Those genes that were shared between three or more GI primary sites also showed significant differential expression in the same direction in other non-GI cancers. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Genome-Wide DNA Methylation Profiling Identifies Differential Methylation in Uninvolved Psoriatic Epidermis
Genome-Wide DNA Methylation Profiling Identifies Differential Methylation in Uninvolved Psoriatic Epidermis Deepti Verma, Anna-Karin Ekman, Cecilia Bivik Eding and Charlotta Enerbäck The self-archived postprint version of this journal article is available at Linköping University Institutional Repository (DiVA): http://urn.kb.se/resolve?urn=urn:nbn:se:liu:diva-147791 N.B.: When citing this work, cite the original publication. Verma, D., Ekman, A., Bivik Eding, C., Enerbäck, C., (2018), Genome-Wide DNA Methylation Profiling Identifies Differential Methylation in Uninvolved Psoriatic Epidermis, Journal of Investigative Dermatology, 138(5), 1088-1093. https://doi.org/10.1016/j.jid.2017.11.036 Original publication available at: https://doi.org/10.1016/j.jid.2017.11.036 Copyright: Elsevier http://www.elsevier.com/ Genome-Wide DNA Methylation Profiling Identifies Differential Methylation in Uninvolved Psoriatic Epidermis Deepti Verma*a, Anna-Karin Ekman*a, Cecilia Bivik Edinga and Charlotta Enerbäcka *Authors contributed equally aIngrid Asp Psoriasis Research Center, Department of Clinical and Experimental Medicine, Division of Dermatology, Linköping University, Linköping, Sweden Corresponding author: Charlotta Enerbäck Ingrid Asp Psoriasis Research Center, Department of Clinical and Experimental Medicine, Linköping University SE-581 85 Linköping, Sweden Phone: +46 10 103 7429 E-mail: [email protected] Short title Differential methylation in psoriasis Abbreviations CGI, CpG island; DMS, differentially methylated site; RRBS, reduced representation bisulphite sequencing Keywords (max 6) psoriasis, epidermis, methylation, Wnt, susceptibility, expression 1 ABSTRACT Psoriasis is a chronic inflammatory skin disease with both local and systemic components. Genome-wide approaches have identified more than 60 psoriasis-susceptibility loci, but genes are estimated to explain only one third of the heritability in psoriasis, suggesting additional, yet unidentified, sources of heritability. -

1 UST College of Science Department of Biological Sciences
UST College of Science Department of Biological Sciences 1 Pharmacogenomics of Myofascial Pain Syndrome An Undergraduate Thesis Submitted to the Department of Biological Sciences College of Science University of Santo Tomas In Partial Fulfillment of the Requirements for the Degree of Bachelor of Science in Biology Jose Marie V. Lazaga Marc Llandro C. Fernandez May 2021 UST College of Science Department of Biological Sciences 2 PANEL APPROVAL SHEET This undergraduate research manuscript entitled: Pharmacogenomics of Myofascial Pain Syndrome prepared and submitted by Jose Marie V. Lazaga and Marc Llandro C. Fernandez, was checked and has complied with the revisions and suggestions requested by panel members after thorough evaluation. This final version of the manuscript is hereby approved and accepted for submission in partial fulfillment of the requirements for the degree of Bachelor of Science in Biology. Noted by: Asst. Prof. Marilyn G. Rimando, PhD Research adviser, Bio/MicroSem 602-603 Approved by: Bio/MicroSem 603 panel member Bio/MicroSem 603 panel member Date: Date: UST College of Science Department of Biological Sciences 3 DECLARATION OF ORIGINALITY We hereby affirm that this submission is our own work and that, to the best of our knowledge and belief, it contains no material previously published or written by another person nor material to which a substantial extent has been accepted for award of any other degree or diploma of a university or other institute of higher learning, except where due acknowledgement is made in the text. We also declare that the intellectual content of this undergraduate research is the product of our work, even though we may have received assistance from others on style, presentation, and language expression. -

Targeted Pharmacological Therapy Restores Β-Cell Function for Diabetes Remission
Targeted pharmacological therapy restores -cell function for diabetes remission Sachs, Stephan; Bastidas-Ponce, Aimée; Tritschler, Sophie; Bakhti, Mostafa; Böttcher, Anika; Sánchez-Garrido, Miguel A; Tarquis-Medina, Marta; Kleinert, Maximilian; Fischer, Katrin; Jall, Sigrid; Harger, Alexandra; Bader, Erik; Roscioni, Sara; Ussar, Siegfried; Feuchtinger, Annette; Yesildag, Burcak; Neelakandhan, Aparna; Jensen, Christine B; Cornu, Marion; Yang, Bin; Finan, Brian; DiMarchi, Richard D; Tschöp, Matthias H; Theis, Fabian J; Hofmann, Susanna M.; Müller, Timo D; Lickert, Heiko Published in: Nature Metabolism DOI: 10.1038/s42255-020-0171-3 Publication date: 2020 Document version Publisher's PDF, also known as Version of record Document license: CC BY Citation for published version (APA): Sachs, S., Bastidas-Ponce, A., Tritschler, S., Bakhti, M., Böttcher, A., Sánchez-Garrido, M. A., Tarquis-Medina, M., Kleinert, M., Fischer, K., Jall, S., Harger, A., Bader, E., Roscioni, S., Ussar, S., Feuchtinger, A., Yesildag, B., Neelakandhan, A., Jensen, C. B., Cornu, M., ... Lickert, H. (2020). Targeted pharmacological therapy restores - cell function for diabetes remission. Nature Metabolism, 2(2), 192-209. https://doi.org/10.1038/s42255-020- 0171-3 Download date: 05. Oct. 2021 ARTICLES https://doi.org/10.1038/s42255-020-0171-3 There are amendments to this paper Targeted pharmacological therapy restores β-cell function for diabetes remission Stephan Sachs1,2,3,4,19, Aimée Bastidas-Ponce1,4,5,6,19, Sophie Tritschler1,4,7,8,19, Mostafa Bakhti 1,4,5, Anika Böttcher1,4,5, Miguel A. Sánchez-Garrido2, Marta Tarquis-Medina1,4,5,6, Maximilian Kleinert2,9, Katrin Fischer2,3, Sigrid Jall2,3, Alexandra Harger2, Erik Bader1, Sara Roscioni1, Siegfried Ussar 4,6,10, Annette Feuchtinger11, Burcak Yesildag12, Aparna Neelakandhan12, Christine B. -
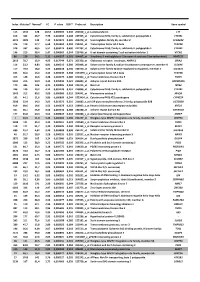
Index Mutated* Normal* FC P Value FDR** Probe Set Description Gene Symbol
Index Mutated* Normal* FC P value FDR** Probe set Description Gene symbol 113 1342 128 10,52 0,000503 0,240 202018_s_at Lactotransferrin LTF 616 362 46,7 7,76 0,003493 0,308 237395_at Cytochrome P450, family 4, subfamily Z, polypeptide 1 CYP4Z1 3073 1009 142 7,10 0,021674 0,385 206378_at Secretoglobin, family 2A, member 2 SCGB2A2 376 119 17,7 6,68 0,001962 0,284 214451_at Transcription factor AP-2 beta TFAP2B 578 337 60,5 5,57 0,003174 0,300 227702_at Cytochrome P450, family 4, subfamily X, polypeptide 1 CYP4X1 146 210 38,4 5,47 0,000669 0,244 219768_at V-set domain containing T cell activation inhibitor 1 VTCN1 86 129 24,2 5,31 0,000327 0,201 204607_at 3-hydroxy-3-methylglutaryl-Coenzyme A synthase 2 (mitochondrial) HMGCS2 2613 78,7 15,9 4,95 0,017744 0,371 205358_at Glutamate receptor, ionotropic, AMPA 2 GRIA2 116 23,2 4,83 4,81 0,000513 0,240 203908_at Solute carrier family 4, sodium bicarbonate cotransporter, member 4 SLC4A4 117 79,4 18,0 4,42 0,000518 0,240 239723_at Solute carrier family 40 (iron-regulated transporter), member 1 SLC40A1 625 56,6 13,0 4,34 0,003549 0,308 1553394_a_atTranscription factor AP-2 beta TFAP2B 413 148 34,6 4,28 0,002175 0,286 240304_s_at Transmembrane channel-like 5 TMC5 5181 216 50,9 4,25 0,039946 0,421 223864_at Ankyrin repeat domain 30A ANKRD30A 179 446 106 4,21 0,000826 0,244 223315_at Netrin 4 NTN4 246 120 29,2 4,12 0,001128 0,251 210096_at Cytochrome P450, family 4, subfamily B, polypeptide 1 CYP4B1 1043 312 80,2 3,89 0,006080 0,319 204041_at Monoamine oxidase B MAOB 192 44,1 11,6 3,80 0,000899 0,244 -

Probabilistic Models for Collecting, Analyzing, and Modeling Expression Data
Probabilistic Models for Collecting, Analyzing, and Modeling Expression Data Hai-Son Phuoc Le May 2013 CMU-ML-13-101 Probabilistic Models for Collecting, Analyzing, and Modeling Expression Data Hai-Son Phuoc Le May 2013 CMU-ML-13-101 Machine Learning Department School of Computer Science Carnegie Mellon University Thesis Committee Ziv Bar-Joseph, Chair Christopher Langmead Roni Rosenfeld Quaid Morris Submitted in partial fulfillment of the requirements for the Degree of Doctor of Philosophy. Copyright @ 2013 Hai-Son Le This research was sponsored by the National Institutes of Health under grant numbers 5U01HL108642 and 1R01GM085022, the National Science Foundation under grant num- bers DBI0448453 and DBI0965316, and the Pittsburgh Life Sciences Greenhouse. The views and conclusions contained in this document are those of the author and should not be interpreted as representing the official policies, either expressed or implied, of any sponsoring institution, the U.S. government or any other entity. Keywords: genomics, gene expression, gene regulation, microarray, RNA-Seq, transcriptomics, error correction, comparative genomics, regulatory networks, cross-species, expression database, Gene Expression Omnibus, GEO, orthologs, microRNA, target prediction, Dirichlet Process, Indian Buffet Process, hidden Markov model, immune response, cancer. To Mom and Dad. i Abstract Advances in genomics allow researchers to measure the complete set of transcripts in cells. These transcripts include messenger RNAs (which encode for proteins) and microRNAs, short RNAs that play an important regulatory role in cellular networks. While this data is a great resource for reconstructing the activity of networks in cells, it also presents several computational challenges. These challenges include the data collection stage which often results in incomplete and noisy measurement, developing methods to integrate several experiments within and across species, and designing methods that can use this data to map the interactions and networks that are activated in specific conditions. -

Identification of Potential Key Genes and Pathway Linked with Sporadic Creutzfeldt-Jakob Disease Based on Integrated Bioinformatics Analyses
medRxiv preprint doi: https://doi.org/10.1101/2020.12.21.20248688; this version posted December 24, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. All rights reserved. No reuse allowed without permission. Identification of potential key genes and pathway linked with sporadic Creutzfeldt-Jakob disease based on integrated bioinformatics analyses Basavaraj Vastrad1, Chanabasayya Vastrad*2 , Iranna Kotturshetti 1. Department of Biochemistry, Basaveshwar College of Pharmacy, Gadag, Karnataka 582103, India. 2. Biostatistics and Bioinformatics, Chanabasava Nilaya, Bharthinagar, Dharwad 580001, Karanataka, India. 3. Department of Ayurveda, Rajiv Gandhi Education Society`s Ayurvedic Medical College, Ron, Karnataka 562209, India. * Chanabasayya Vastrad [email protected] Ph: +919480073398 Chanabasava Nilaya, Bharthinagar, Dharwad 580001 , Karanataka, India NOTE: This preprint reports new research that has not been certified by peer review and should not be used to guide clinical practice. medRxiv preprint doi: https://doi.org/10.1101/2020.12.21.20248688; this version posted December 24, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. All rights reserved. No reuse allowed without permission. Abstract Sporadic Creutzfeldt-Jakob disease (sCJD) is neurodegenerative disease also called prion disease linked with poor prognosis. The aim of the current study was to illuminate the underlying molecular mechanisms of sCJD. The mRNA microarray dataset GSE124571 was downloaded from the Gene Expression Omnibus database. Differentially expressed genes (DEGs) were screened. -

(P -Value<0.05, Fold Change≥1.4), 4 Vs. 0 Gy Irradiation
Table S1: Significant differentially expressed genes (P -Value<0.05, Fold Change≥1.4), 4 vs. 0 Gy irradiation Genbank Fold Change P -Value Gene Symbol Description Accession Q9F8M7_CARHY (Q9F8M7) DTDP-glucose 4,6-dehydratase (Fragment), partial (9%) 6.70 0.017399678 THC2699065 [THC2719287] 5.53 0.003379195 BC013657 BC013657 Homo sapiens cDNA clone IMAGE:4152983, partial cds. [BC013657] 5.10 0.024641735 THC2750781 Ciliary dynein heavy chain 5 (Axonemal beta dynein heavy chain 5) (HL1). 4.07 0.04353262 DNAH5 [Source:Uniprot/SWISSPROT;Acc:Q8TE73] [ENST00000382416] 3.81 0.002855909 NM_145263 SPATA18 Homo sapiens spermatogenesis associated 18 homolog (rat) (SPATA18), mRNA [NM_145263] AA418814 zw01a02.s1 Soares_NhHMPu_S1 Homo sapiens cDNA clone IMAGE:767978 3', 3.69 0.03203913 AA418814 AA418814 mRNA sequence [AA418814] AL356953 leucine-rich repeat-containing G protein-coupled receptor 6 {Homo sapiens} (exp=0; 3.63 0.0277936 THC2705989 wgp=1; cg=0), partial (4%) [THC2752981] AA484677 ne64a07.s1 NCI_CGAP_Alv1 Homo sapiens cDNA clone IMAGE:909012, mRNA 3.63 0.027098073 AA484677 AA484677 sequence [AA484677] oe06h09.s1 NCI_CGAP_Ov2 Homo sapiens cDNA clone IMAGE:1385153, mRNA sequence 3.48 0.04468495 AA837799 AA837799 [AA837799] Homo sapiens hypothetical protein LOC340109, mRNA (cDNA clone IMAGE:5578073), partial 3.27 0.031178378 BC039509 LOC643401 cds. [BC039509] Homo sapiens Fas (TNF receptor superfamily, member 6) (FAS), transcript variant 1, mRNA 3.24 0.022156298 NM_000043 FAS [NM_000043] 3.20 0.021043295 A_32_P125056 BF803942 CM2-CI0135-021100-477-g08 CI0135 Homo sapiens cDNA, mRNA sequence 3.04 0.043389246 BF803942 BF803942 [BF803942] 3.03 0.002430239 NM_015920 RPS27L Homo sapiens ribosomal protein S27-like (RPS27L), mRNA [NM_015920] Homo sapiens tumor necrosis factor receptor superfamily, member 10c, decoy without an 2.98 0.021202829 NM_003841 TNFRSF10C intracellular domain (TNFRSF10C), mRNA [NM_003841] 2.97 0.03243901 AB002384 C6orf32 Homo sapiens mRNA for KIAA0386 gene, partial cds. -

Hippo and Sonic Hedgehog Signalling Pathway Modulation of Human Urothelial Tissue Homeostasis
Hippo and Sonic Hedgehog signalling pathway modulation of human urothelial tissue homeostasis Thomas Crighton PhD University of York Department of Biology November 2020 Abstract The urinary tract is lined by a barrier-forming, mitotically-quiescent urothelium, which retains the ability to regenerate following injury. Regulation of tissue homeostasis by Hippo and Sonic Hedgehog signalling has previously been implicated in various mammalian epithelia, but limited evidence exists as to their role in adult human urothelial physiology. Focussing on the Hippo pathway, the aims of this thesis were to characterise expression of said pathways in urothelium, determine what role the pathways have in regulating urothelial phenotype, and investigate whether the pathways are implicated in muscle-invasive bladder cancer (MIBC). These aims were assessed using a cell culture paradigm of Normal Human Urothelial (NHU) cells that can be manipulated in vitro to represent different differentiated phenotypes, alongside MIBC cell lines and The Cancer Genome Atlas resource. Transcriptomic analysis of NHU cells identified a significant induction of VGLL1, a poorly understood regulator of Hippo signalling, in differentiated cells. Activation of upstream transcription factors PPARγ and GATA3 and/or blockade of active EGFR/RAS/RAF/MEK/ERK signalling were identified as mechanisms which induce VGLL1 expression in NHU cells. Ectopic overexpression of VGLL1 in undifferentiated NHU cells and MIBC cell line T24 resulted in significantly reduced proliferation. Conversely, knockdown of VGLL1 in differentiated NHU cells significantly reduced barrier tightness in an unwounded state, while inhibiting regeneration and increasing cell cycle activation in scratch-wounded cultures. A signalling pathway previously observed to be inhibited by VGLL1 function, YAP/TAZ, was unaffected by VGLL1 manipulation. -

Gene Expression in the Brain of a Migratory Songbird During Breeding and Migration
Boss et al. Movement Ecology (2016) 4:4 DOI 10.1186/s40462-016-0069-6 RESEARCH Open Access Gene expression in the brain of a migratory songbird during breeding and migration John Boss1,2, Miriam Liedvogel3,6, Max Lundberg3, Peter Olsson4, Nils Reischke3, Sara Naurin3, Susanne Åkesson5, Dennis Hasselquist3, Anthony Wright1, Mats Grahn1 and Staffan Bensch3* Abstract Background: We still have limited knowledge about the underlying genetic mechanisms that enable migrating species of birds to navigate the globe. Here we make an attempt to get insight into the genetic architecture controlling this complex innate behaviour. We contrast the gene expression profiles of two closely related songbird subspecies with divergent migratory phenotypes. In addition to comparing differences in migratory strategy we include a temporal component and contrast patterns between breeding adults and autumn migrating juvenile birds of both subspecies. The two willow warbler subspecies, Phylloscopus trochilus trochilus and P. t. acredula, are remarkably similar both in phenotype and genotype and have a narrow contact zone in central Scandinavia. Here we used a microarray gene chip representing 23,136 expressed sequence tags (ESTs) from the zebra finch Taeniopygia guttata to identify mRNA level differences in willow warbler brain tissue in relation to subspecies and season. Results: Out of the 22,109 EST probe sets that remained after filtering poorly binding probes, we found 11,898 (51.8 %) probe sets that could be reliably and uniquely matched to a total of 6,758 orthologous zebra finch genes. The two subspecies showed very similar levels of gene expression with less than 0.1 % of the probe sets being significantly differentially expressed.