Neuronal Mitophagy: Lessons from a Pathway Linked to Parkinson's
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
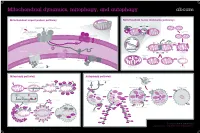
Mitochondrial Dynamics, Mitophagy, and Autophagy
Mitochondrial dynamics, mitophagy, and autophagy Mitochondrial import protein pathway Mitochondrial fusion and fission pathways iΔΨm, (OXPHOSh) Ca2+ Fusion ++++ (Cell differentiationi) N Internal signal β-barrel outer Opa1 SH ER sequence SH membrane precursors Mfn1/2 SH SH GTP GTP C Inner membrane and GTP GTP Outer membrane Matrix carrier precursors 22 70 GTP fusion (N-terminal presequences) 35 GTP 20 37 Tom complex 5 Sam complex Opa1 Cytosol Outer 6 Sam50 7 Mdm10 membrane DRP1 Fis1 Inner membrane fusion Tim8-Tim13 Mim 1 Tom40 Bax/Bak apoptosis Outer membrane HR2 regions S-S Tim9-Tim10 S-S Inner Erv1 95 A 54 mitochondrial S-S Reorganization Mia40 space 21 Fission sequestration 50 18 Tim22 Translocation Depolarization to meet iΔΨm ATP needs Tim23 KREBS Cycle NADH FADH2 17 Oxa1 Tim 22 complex Mia Mba1 Mdm I ATP 44 38 H+ II 17 H+ Inner membrane 16 +++ III ADP - Pi 18 Ribosome Oxa H+ IV mtHsp70 H+ ATP +++ Mge1 MPP Healthy mitochondrion Mitophagy Matrix Tim 23 complex Mitophagy pathway Autophagy pathway Lysosomal hydroiase Lysosome Hypoxia, Nutrient/Growth Rapamycin Starvation condition Ubiquitin Cytosol Parkin factor deprivation Lamp Stress Damaged mitochondrion Parkin Parkin iΔΨm Parkin MARF PINK1 PINK1 PINK1 Mfn1 VDAC1 Parkin AMPK mTOR Mfn2 PINK1 PINK1 Phagophore Autophagosome Autophagolyosome PARL LC3-II PINK1-L PINK1-S Parkin P FIS1 ATG13 ULK LC3-II Membrane PINK1 PINK1 FIP200 PINK1 MIRO2 P Bak PINK1 MIRO1 Parkin Parkin Parkin Cytoplasmic Cytosol proLC3 macromolecule LC3-II Atg4B Atg7/LC3-I Atg5/Atg12/Atg16 Organelle Atg9 PE Atg18 Atg2 -

Involvement of Impaired Autophagy and Mitophagy in Neuro-2A Cell
www.nature.com/scientificreports OPEN Involvement of impaired autophagy and mitophagy in Neuro-2a cell damage under hypoxic and/or high- Received: 19 May 2017 Accepted: 15 January 2018 glucose conditions Published: xx xx xxxx Yufei Song, Yu Du, Wenying Zou, Yan Luo, Xiaojie Zhang & Jianliang Fu Chronic cerebral hypoperfusion (CCH) plays an insidious role in the development of cognitive impairment. Considerable evidence suggests that Diabetes Mellitus (DM) as a vascular risk factor may exacerbate CCH and is closely related to cognitive decline. Dysregulation of autophagy is known to be associated with the pathogenesis of neurodegenerative diseases such as Alzheimer’s disease. To elucidate the role of autophagy in CCH- and/or DM-related pathogenesis, mouse neuroblastoma Neuro-2a cells were exposed to hypoxia and/or high glucose for 48 h, mimicking CCH complicated with DM pathologies. Chronic hypoxia reduced cell proliferation and increased levels of cleaved caspase-3, whereas high glucose had no obvious synergistic toxic efect. Accumulation of autophagic vacuoles under hypoxia may be due to both autophagy impairment and induction, with the former accounting for Neuro-2a cell death. Additionally, aberrant accumulation of mitochondria in Neuro-2a cells may be attributed to insufcient BNIP3-mediated mitophagy due to poor interaction between BNIP3 and LC3-II. Despite the lack of a signifcant cytotoxic efect of high glucose under our experimental conditions, our data indicated for the frst time that impaired autophagy degradation and inefcient BNIP3-mediated mitophagy may constitute mechanisms underlying neuronal cell damage during chronic hypoxia. Chronic cerebral hypoperfusion (CCH) is a normal process related to ageing that likely contributes to age-related memory loss1. -

Mitoq Inhibits Hepatic Stellate Cell Activation and Liver Fibrosis by Enhancing PINK1/Parkin-Mediated Mitophagy
MitoQ Inhibits Hepatic Stellate Cell Activation and Liver Fibrosis by Enhancing PINK1/parkin-mediated Mitophagy Shi-Ying Dou Second Hospital of Hebei Medical University Jiu-Na Zhang Second Hospital of Hebei Medical University Xiao-Li Xie Second Hospital of Hebei Medical University Ting Liu Second Hospital of Hebei Medical University Jun-Li Hu Second Hospital of Hebei Medical University Xiao-Yu Jiang Second Hospital of Hebei Medical University Miao-Miao Wang Second Hospital of Hebei Medical University Hui-Qing Jiang ( [email protected] ) Second Hospital of Hebei Medical University https://orcid.org/0000-0002-5235-1457 Research Article Keywords: Liver brosis, hepatic stellate cell, ubiquinone, PINK1 mitophagy Posted Date: March 30th, 2021 DOI: https://doi.org/10.21203/rs.3.rs-344963/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/18 Abstract Mitophagy plays an important role in the activation of hepatic stellate cells (HSCs). Mitochondria- targeted ubiquinone (MitoQ) is a mitochondria-targeted antioxidant that reduces the production of intracellular reactive oxygen species (ROS). However, its relationship with mitophagy remains unclear. This study evaluated mitophagy during HSC activation and the effects of MitoQ on mitophagy in cell culture and in an animal model of the activation of HSCs. We found that MitoQ reduced the activation of HSCs and alleviated hepatic brosis. While activation of primary HSCs or LX-2 cells was associated with reduced PINK1/parkin-mediated mitophagy, MitoQ reduced intracellular ROS levels, enhanced PINK1/parkin-mediated mitophagy, and inhibited the activation of HSCs. After knocking down the key mitophagy-related protein, PINK1, in LX-2 cells to block mitophagy, MitoQ intervention failed to inhibit HSC activation. -

Gp78 E3 Ubiquitin Ligase Mediates Both Basal and Damage-Induced Mitophagy
bioRxiv preprint doi: https://doi.org/10.1101/407593; this version posted September 3, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Gp78 E3 ubiquitin ligase mediates both basal and damage-induced mitophagy Bharat Joshi, Yahya Mohammadzadeh, Guang Gao and Ivan R. Nabi Department of Cellular and Physiological Sciences, Life Sciences Institute, University of British Columbia, Vancouver, BC V6T 1Z3, Canada #Running title: Gp78 control of mitophagy §To whom correspondence should be addressed: Ivan R. Nabi, Department of Cellular and Physiological Sciences, Life Sciences Institute, University of British Columbia, 2350 Health Sciences Mall, Vancouver, BC V6T 1Z3 Canada. Tel: +1-(604) 822-7000 E-mail: [email protected] Key words: Gp78 ubiquitin ligase; mitochondria; autophagy; PINK1; Parkin bioRxiv preprint doi: https://doi.org/10.1101/407593; this version posted September 3, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract Mitophagy, the elimination of mitochondria by the autophagy machinery, evolved to monitor mitochondrial health and maintain mitochondrial integrity. PINK1 is a sensor of mitochondrial health that recruits Parkin and other mitophagy-inducing ubiquitin ligases to depolarized mitochondria. However, mechanisms underlying mitophagic control of mitochondrial homeostasis, basal mitophagy, remain poorly understood. The Gp78 E3 ubiquitin ligase, an endoplasmic reticulum membrane protein, induces mitochondrial fission, endoplasmic reticulum- mitochondria contacts and mitophagy of depolarized mitochondria. CRISPR/Cas9 knockout of Gp78 in HT-1080 fibrosarcoma cells results in reduced ER-mitochondria contacts, increased mitochondrial volume and resistance to CCCP-induced mitophagy. -

Mitochondrial Physiology Within Myelinated Axons in Health and Disease : an Energetic Interplay Between Counterparts Gerben Van Hameren
Mitochondrial physiology within myelinated axons in health and disease : an energetic interplay between counterparts Gerben Van Hameren To cite this version: Gerben Van Hameren. Mitochondrial physiology within myelinated axons in health and disease : an energetic interplay between counterparts. Human health and pathology. Université Montpellier, 2018. English. NNT : 2018MONTT084. tel-02053421 HAL Id: tel-02053421 https://tel.archives-ouvertes.fr/tel-02053421 Submitted on 1 Mar 2019 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. THÈSE POUR OBTENIR LE GRADE DE DOCTEUR DE L’UNIVERSITÉ DE M ONTPELLIER En Biologie Santé École doctorale CBS2 Institut des Neurosciences de Montpellier MITOCHONDRIAL PHYSIOLOGY WITHIN MYELINATED AXONS IN HEALTH AND DISEASE AN ENERGETIC INTERPLAY BETWEEN COUNTERPARTS Présentée par Gerben van Hameren Le 23 Novembre 2018 Sous la direction de Dr. Nicolas Tricaud Devant le jury composé de Prof. Pascale Belenguer, Centre de Recherches sur la Cognition Animale Toulouse Professeur d’université Dr. Guy Lenaers, Mitochondrial Medicine Research Centre Angers Directeur de recherche Dr. Don Mahad, University of Edinburgh Senior clinical lecturer Invité Dr. Marie-Luce Vignais, Institute for Regenerative Medicine & Biotherapy, Montpellier Chargé de recherche 0 Table of contents Prologue ................................................................................................................................................. -

In Vivo Real-Time Dynamics of ATP and ROS Production in Axonal
Hameren et al. Acta Neuropathologica Communications (2019) 7:86 https://doi.org/10.1186/s40478-019-0740-4 RESEARCH Open Access In vivo real-time dynamics of ATP and ROS production in axonal mitochondria show decoupling in mouse models of peripheral neuropathies Gerben van Hameren1* , Graham Campbell1, Marie Deck1, Jade Berthelot1, Benoit Gautier1, Patrice Quintana1, Roman Chrast2,3 and Nicolas Tricaud1* Abstract Mitochondria are critical for the function and maintenance of myelinated axons notably through Adenosine triphosphate (ATP) production. A direct by-product of this ATP production is reactive oxygen species (ROS), which are highly deleterious for neurons. While ATP shortage and ROS levels increase are involved in several neurodegenerative diseases, it is still unclear whether the real-time dynamics of both ATP and ROS production in axonal mitochondria are altered by axonal or demyelinating neuropathies. To answer this question, we imaged and quantified mitochondrial ATP and hydrogen peroxide (H2O2) in resting or stimulated peripheral nerve myelinated axons in vivo, using genetically-encoded fluorescent probes, two-photon time-lapse and CARS imaging. We found that ATP and H2O2 productions are intrinsically higher in nodes of Ranvier even in resting conditions. Axonal firing increased both ATP and H2O2 productions but with different dynamics: ROS production peaked shortly and transiently after the stimulation while ATP production increased gradually for a longer period of time. In neuropathic MFN2R94Q mice, mimicking Charcot-Marie-Tooth 2A disease, defective mitochondria failed to upregulate ATP production following axonal activity. However, elevated H2O2 production was largely sustained. Finally, inducing demyelination with lysophosphatidylcholine resulted in a reduced level of ATP while H2O2 level soared. -

Roles of Sigma-1 Receptors on Mitochondrial Functions Relevant to Neurodegenerative Diseases Tzu-Yu Weng1,2, Shang-Yi Anne Tsai1 and Tsung-Ping Su1*
Weng et al. Journal of Biomedical Science (2017) 24:74 DOI 10.1186/s12929-017-0380-6 REVIEW Open Access Roles of sigma-1 receptors on mitochondrial functions relevant to neurodegenerative diseases Tzu-Yu Weng1,2, Shang-Yi Anne Tsai1 and Tsung-Ping Su1* Abstract The sigma-1 receptor (Sig-1R) is a chaperone that resides mainly at the mitochondrion-associated endoplasmic reticulum (ER) membrane (called the MAMs) and acts as a dynamic pluripotent modulator in living systems. At the MAM, the Sig-1R is known to play a role in regulating the Ca2+ signaling between ER and mitochondria and in maintaining the structural integrity of the MAM. The MAM serves as bridges between ER and mitochondria regulating multiple functions such as Ca2+ transfer, energy exchange, lipid synthesis and transports, and protein folding that are pivotal to cell survival and defense. Recently, emerging evidences indicate that the MAM is critical in maintaining neuronal homeostasis. Thus, given the specific localization of the Sig-1R at the MAM, we highlight and propose that the direct or indirect regulations of the Sig-1R on mitochondrial functions may relate to neurodegenerative diseases including Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD) and amyotrophic lateral sclerosis (ALS). In addition, the promising use of Sig-1R ligands to rescue mitochondrial dysfunction-induced neurodegeneration is addressed. Keywords: Sigma-1 receptor, Mitochondria, Mitochondrion-associated ER membrane (MAM), Neurodegenerative disorders Background also regulates Ca2+ influx by attenuating the coupling of The sigma-1 receptor (Sig-1R) is an endoplasmic the ER Ca2+ sensor STIM1 to Orai1 [3]. -

Astrocytic Reactivity Triggered by Defective Mitophagy Activates NF-Kb Signaling and Causes Neurotoxicity in Frontotemporal Dementia Type 3
Astrocytic Reactivity Triggered by defective Mitophagy Activates NF-kB Signaling and Causes Neurotoxicity in Frontotemporal Dementia Type 3 Abinaya chandrasekaran Kobenhavns Universitet Katarina Stoklund Dittlau KU Leuven: Katholieke Universiteit Leuven Giulia Corsi Kobenhavns Universitet Henriette Haukedal Kobenhavns Universitet Nadezhda Doncheva Kobenhavns Universitet Sarayu Ramakrishna Institute for Stem Cell Science and Regenerative Medicine Scheetal Ambardar Institute for Stem Cell Science and Regenerative Medicine Claudia Salcedo Kobenhavns Universitet Sissel Schmidt Odense University: Syddansk Universitet Yu Zhang Lunds Universitet Susanna Cirera Kobenhavns Universitet Benjamin Schmid Bioneer A/S Troels Tolstrup Nielsen University of Copenhagen Jørgen Erik Nielsen University of Copenhagen Miriam Kolko Kobenhavns Universitet Julianna Kobolak Page 1/11 BioTalentum Ltd Andras Dinnyes BioTalentum Ltd Poul Hyttel Kobenhavns Universitet Dasaradhi Palakodeti inStem: Institute for Stem Cell Science and Regenerative Medicine Jan Gorodkin Kobenhavns Universitet Ravi Muddashetty inStem: Institute for Stem Cell Science and Regenerative Medicine Morten Meyer Odense University: Syddansk Universitet Blanca Irene Aldana Kobenhavns Universitet Kristine Freude ( [email protected] ) Kobenhavns Universitet https://orcid.org/0000-0001-9480-2386 Research article Keywords: CHMP2B FTD3, hiPSC-derived astrocytes, autophagy, mitochondria, cytokines Posted Date: March 16th, 2021 DOI: https://doi.org/10.21203/rs.3.rs-310167/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 2/11 Abstract Background: Frontotemporal dementia type 3 (FTD3) caused by a point mutation in the charged multivesicular body protein 2B (CHMP2B), affects mitochondrial ultrastructure and function as well as endosomal-lysosomal fusion in neurons. However, there is a critical knowledge gap in understanding how mutations in CHMP2B affect astrocytes. -

Plant Mitophagy in Comparison to Mammals: What Is Still Missing?
International Journal of Molecular Sciences Review Plant Mitophagy in Comparison to Mammals: What Is Still Missing? Kaike Ren, Lanlan Feng, Shuangli Sun and Xiaohong Zhuang * Centre for Cell and Developmental Biology, State Key Laboratory of Agrobiotechnology, School of Life Sciences, The Chinese University of Hong Kong, Hong Kong, China; [email protected] (K.R.); [email protected] (L.F.); [email protected] (S.S.) * Correspondence: [email protected] Abstract: Mitochondrial homeostasis refers to the balance of mitochondrial number and quality in a cell. It is maintained by mitochondrial biogenesis, mitochondrial fusion/fission, and the clearance of unwanted/damaged mitochondria. Mitophagy represents a selective form of autophagy by seques- tration of the potentially harmful mitochondrial materials into a double-membrane autophagosome, thus preventing the release of death inducers, which can trigger programmed cell death (PCD). Recent advances have also unveiled a close interconnection between mitophagy and mitochondrial dynamics, as well as PCD in both mammalian and plant cells. In this review, we will summarize and discuss recent findings on the interplay between mitophagy and mitochondrial dynamics, with a focus on the molecular evidence for mitophagy crosstalk with mitochondrial dynamics and PCD. Keywords: mitochondrial homeostasis; mitophagy; mitochondrial fission; mitochondrial fusion; programmed cell death 1. Introduction Citation: Ren, K.; Feng, L.; Sun, S.; The mitochondrion is an essential organelle conserved in eukaryotic organisms, which Zhuang, X. Plant Mitophagy in conducts several key biological functions. The primary function of mitochondria is to Comparison to Mammals: What Is manufacture ATP, the major energy resource of the cells, through oxidative respiration Still Missing?. -

Mdm2 Enhances Ligase Activity of Parkin and Facilitates Mitophagy Seunghyi Kook1,2, Xuanzhi Zhan1,3, Kimberly Thibeault1, Mohamed R
www.nature.com/scientificreports OPEN Mdm2 enhances ligase activity of parkin and facilitates mitophagy Seunghyi Kook1,2, Xuanzhi Zhan1,3, Kimberly Thibeault1, Mohamed R. Ahmed1,4, Vsevolod V. Gurevich 1 & Eugenia V. Gurevich1* Loss-of-function mutations in the E3 ubiquitin ligase parkin have been implicated in the death of dopaminergic neurons in the substantia nigra, which is the root cause of dopamine defcit in the striatum in Parkinson's disease. Parkin ubiquitinates proteins on mitochondria that lost membrane potential, promoting the elimination of damaged mitochondria. Neuroprotective activity of parkin has been linked to its critical role in the mitochondria maintenance. Here we report a novel regulatory mechanism: another E3 ubiquitin ligase Mdm2 directly binds parkin and enhances its enzymatic activity in vitro and in intact cells. Mdm2 translocates to damaged mitochondria independently of parkin, enhances parkin-dependent ubiquitination of the outer mitochondria membrane protein mitofusin1. Mdm2 facilitates and its knockdown reduces parkin-dependent mitophagy. Thus, ubiquitously expressed Mdm2 might enhance cytoprotective parkin activity. The data suggest that parkin activation by Mdm2 could be targeted to increase its neuroprotective functions, which has implications for anti- parkinsonian therapy. Parkin was frst identifed through its association with autosomal recessive juvenile parkinsonism, a familial form of Parkinson's disease (PD) with early onset1. Loss-of-function mutations in parkin gene, including exon deletions and rearrangements, as well as nonsense and missense mutations, are commonly associated not only with familial, but also with apparently sporadic early onset PD2–5. Functionally, parkin is an E3 ubiquitin ligase6. Initially, the studies of parkin function in PD concentrated on the search for parkin substrates and the role of parkin loss in the dysfunction of the ubiquitin-proteasome system (UBS), which appears to play a prominent role in the PD-related neurodegeneration7. -

Functional Characterization of Novel Rhot1 Variants, Which Are Associated with Parkinson’S Disease
FUNCTIONAL CHARACTERIZATION OF NOVEL RHOT1 VARIANTS, WHICH ARE ASSOCIATED WITH PARKINSON’S DISEASE DISSERTATION zur Erlangung des Grades eines DOKTORS DER NATURWISSENSCHAFTEN DER MATHEMATISCH-NATURWISSENSCHAFTLICHEN FAKULTÄT und DER MEDIZINISCHEN FAKULTÄT DER EBERHARD-KARLS-UNIVERSITÄT TÜBINGEN vorgelegt von DAJANA GROßMANN aus Wismar, Deutschland Mai 2016 II PhD-FSTC-2016-15 The Faculty of Sciences, Technology and Communication The Faculty of Science and Medicine and The Graduate Training Centre of Neuroscience DISSERTATION Defense held on 13/05/2016 in Luxembourg to obtain the degree of DOCTEUR DE L’UNIVERSITÉ DU LUXEMBOURG EN BIOLOGIE AND DOKTOR DER EBERHARD-KARLS-UNIVERISTÄT TÜBINGEN IN NATURWISSENSCHAFTEN by Dajana GROßMANN Born on 14 August 1985 in Wismar (Germany) FUNCTIONAL CHARACTERIZATION OF NOVEL RHOT1 VARIANTS, WHICH ARE ASSOCIATED WITH PARKINSON’S DISEASE. III IV Date of oral exam: 13th of May 2016 President of the University of Tübingen: Prof. Dr. Bernd Engler …………………………………… Chairmen of the Doctorate Board of the University of Tübingen: Prof. Dr. Bernd Wissinger …………………………………… Dekan der Math.-Nat. Fakultät: Prof. Dr. W. Rosenstiel …………………………………… Dekan der Medizinischen Fakultät: Prof. Dr. I. B. Autenrieth .................................................. President of the University of Luxembourg: Prof. Dr. Rainer Klump …………………………………… Supervisor from Luxembourg: Prof. Dr. Rejko Krüger …………………………………… Supervisor from Tübingen: Prof. Dr. Olaf Rieß …………………………………… Dissertation Defence Committee: Committee members: Dr. Alexander -

Mitochondria-Associated Membranes (Mams) Are Involved in Bax Mitochondrial Localization and Cytochrome C Release
bioRxiv preprint doi: https://doi.org/10.1101/443606; this version posted October 15, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Mitochondria-Associated Membranes (MAMs) are involved in Bax mitochondrial localization and cytochrome c release Alexandre Légiot, Claire Céré, Thibaud Dupoiron, Mohamed Kaabouni, Stéphen Manon1 Institut de Biochimie et de Génétique Cellulaires, UMR 5095 CNRS & Université de Bordeaux, Campus Carreire, CS61390, 1 Rue Camille Saint-Saëns, 33077 Bordeaux, France 1: correspondence to [email protected] Abstract The distribution of the pro-apoptotic protein Bax in the outer mitochondrial membrane (OMM) is a central point of regulation of apoptosis. It is now widely recognized that parts of the endoplasmic reticulum (ER) are closely associated to the OMM, and are actively involved in different signalling processes. We adressed a possible role of these domains, called Mitochondria-Associated Membranes (MAMs) in Bax localization and fonction, by expressing the human protein in a yeast mutant deleted of MDM34, a ERMES component (ER-Mitochondria Encounter Structure). By affecting MAMs stability, the deletion of MDM34 altered Bax mitochondrial localization, and decreased its capacity to release cytochrome c. Furthermore, the deletion of MDM34 decreased the size of an uncompletely released, MAMs-associated pool of cytochrome c. Introduction Apoptosis, the major programmed cell death pathway in animals, plays a central role during development, and along the whole life, by mediating the elimination of dispensable or potentially dangerous cells. Apoptosis is also involved in the response of cells to toxic molecules, such as anti- tumoral drugs.