Supporting Materials
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Influencers on Thyroid Cancer Onset: Molecular Genetic Basis
G C A T T A C G G C A T genes Review Influencers on Thyroid Cancer Onset: Molecular Genetic Basis Berta Luzón-Toro 1,2, Raquel María Fernández 1,2, Leticia Villalba-Benito 1,2, Ana Torroglosa 1,2, Guillermo Antiñolo 1,2 and Salud Borrego 1,2,* 1 Department of Maternofetal Medicine, Genetics and Reproduction, Institute of Biomedicine of Seville (IBIS), University Hospital Virgen del Rocío/CSIC/University of Seville, 41013 Seville, Spain; [email protected] (B.L.-T.); [email protected] (R.M.F.); [email protected] (L.V.-B.); [email protected] (A.T.); [email protected] (G.A.) 2 Centre for Biomedical Network Research on Rare Diseases (CIBERER), 41013 Seville, Spain * Correspondence: [email protected]; Tel.: +34-955-012641 Received: 3 September 2019; Accepted: 6 November 2019; Published: 8 November 2019 Abstract: Thyroid cancer, a cancerous tumor or growth located within the thyroid gland, is the most common endocrine cancer. It is one of the few cancers whereby incidence rates have increased in recent years. It occurs in all age groups, from children through to seniors. Most studies are focused on dissecting its genetic basis, since our current knowledge of the genetic background of the different forms of thyroid cancer is far from complete, which poses a challenge for diagnosis and prognosis of the disease. In this review, we describe prevailing advances and update our understanding of the molecular genetics of thyroid cancer, focusing on the main genes related with the pathology, including the different noncoding RNAs associated with the disease. -

Genotyping of Breech Flystrike Resource – Update
Project No: ON-00515 Contract No: PO4500010753 AWI Project Manager: Bridget Peachey Contractor Name: CSIRO Agriculture and Food Prepared by: Dr Sonja Dominik Publication Date: July 2019 Genotyping of breech flystrike resource – update Published by Australian Wool Innovation Limited, Level 6, 68 Harrington Street, THE ROCKS, NSW, 2000 This publication should only be used as a general aid and is not a substitute for specific advice. To the extent permitted by law, we exclude all liability for loss or damage arising from the use of the information in this publication. AWI invests in research, development, innovation and marketing activities along the global supply chain for Australian wool. AWI is grateful for its funding, which is primarily provided by Australian woolgrowers through a wool levy and by the Australian Government which provides a matching contribution for eligible R&D activities © 2019 Australian Wool Innovation Ltd. All rights reserved. Contents Executive Summary .................................................................................................................... 3 1 Introduction/Hypothesis .................................................................................................... 5 2 Literature Review ............................................................................................................... 6 3 Project Objectives .............................................................................................................. 8 4 Success in Achieving Objectives ........................................................................................ -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Repeated BCG Treatment of Mouse Bladder Selectively Stimulates Small
BMC Cancer BioMed Central Research article Open Access Repeated BCG treatment of mouse bladder selectively stimulates small GTPases and HLA antigens and inhibits single-spanning uroplakins Marcia R Saban1, Helen L Hellmich2, Cindy Simpson1, Carole A Davis1, Mark L Lang3, Michael A Ihnat4, Michael A O'Donnell5, Xue-Ru Wu6 and Ricardo Saban*1 Address: 1Department of Physiology, The University Oklahoma Health Sciences Center, Oklahoma City, USA, 2Department of Anesthesiology, University of Texas Medical Branch, Galveston, USA, 3Department of Microbiology and Immunology The University Oklahoma Health Sciences Center, Oklahoma City, OK 73104, USA, 4Department of Cell Biology, The University Oklahoma Health Sciences Center, Oklahoma City, OK 73104, USA, 5Department of Urology, University of Iowa, UI Hospitals and Clinics, Iowa City, Iowa 52242-1089, USA and 6Department of Urology, New York University, School of Medicine, New York, NY 10016, USA Email: Marcia R Saban - [email protected]; Helen L Hellmich - [email protected]; Cindy Simpson - [email protected]; Carole A Davis - [email protected]; Mark L Lang - [email protected]; Michael A Ihnat - [email protected]; Michael A O'Donnell - [email protected]; Xue-Ru Wu - [email protected]; Ricardo Saban* - [email protected] * Corresponding author Published: 2 November 2007 Received: 29 August 2007 Accepted: 2 November 2007 BMC Cancer 2007, 7:204 doi:10.1186/1471-2407-7-204 This article is available from: http://www.biomedcentral.com/1471-2407/7/204 © 2007 Saban et al; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. -

Hippo and Sonic Hedgehog Signalling Pathway Modulation of Human Urothelial Tissue Homeostasis
Hippo and Sonic Hedgehog signalling pathway modulation of human urothelial tissue homeostasis Thomas Crighton PhD University of York Department of Biology November 2020 Abstract The urinary tract is lined by a barrier-forming, mitotically-quiescent urothelium, which retains the ability to regenerate following injury. Regulation of tissue homeostasis by Hippo and Sonic Hedgehog signalling has previously been implicated in various mammalian epithelia, but limited evidence exists as to their role in adult human urothelial physiology. Focussing on the Hippo pathway, the aims of this thesis were to characterise expression of said pathways in urothelium, determine what role the pathways have in regulating urothelial phenotype, and investigate whether the pathways are implicated in muscle-invasive bladder cancer (MIBC). These aims were assessed using a cell culture paradigm of Normal Human Urothelial (NHU) cells that can be manipulated in vitro to represent different differentiated phenotypes, alongside MIBC cell lines and The Cancer Genome Atlas resource. Transcriptomic analysis of NHU cells identified a significant induction of VGLL1, a poorly understood regulator of Hippo signalling, in differentiated cells. Activation of upstream transcription factors PPARγ and GATA3 and/or blockade of active EGFR/RAS/RAF/MEK/ERK signalling were identified as mechanisms which induce VGLL1 expression in NHU cells. Ectopic overexpression of VGLL1 in undifferentiated NHU cells and MIBC cell line T24 resulted in significantly reduced proliferation. Conversely, knockdown of VGLL1 in differentiated NHU cells significantly reduced barrier tightness in an unwounded state, while inhibiting regeneration and increasing cell cycle activation in scratch-wounded cultures. A signalling pathway previously observed to be inhibited by VGLL1 function, YAP/TAZ, was unaffected by VGLL1 manipulation. -

Genes Involved in Differentiation, Stem Cell Renewal, and Tumorigenesis Are Modulated in Telomerase- Immortalized Human Urothelial Cells
Genes Involved in Differentiation, Stem Cell Renewal, and Tumorigenesis Are Modulated in Telomerase- Immortalized Human Urothelial Cells Emma J. Chapman,1 Gavin Kelly,2 and Margaret A. Knowles1 1Cancer Research UK Clinical Centre, Leeds Institute of Molecular Medicine, St. James’s University Hospital, Leeds, United Kingdom and 2Bioinformatics and Biostatistics Service, Cancer Research UK, London Research Institute, Lincoln’s Inn Fields Laboratories, London, United Kingdom Abstract non–telomere effects of telomerase and provides further The expression of hTERT, the catalytic subunit of rationale for the use of telomerase inhibitors in UC. telomerase, immortalizes normal human urothelial cells (Mol Cancer Res 2008;6(7):1154–68) (NHUC). Expression of a modified hTERT, without the ability to act in telomere maintenance, did not Introduction immortalize NHUC, confirming that effects at telomeres The primary and well-documented role of telomerase is as are required for urothelial immortalization. Previous a reverse transcriptase that acts in the maintenance of studies indicate that inhibition of telomerase has an telomere length and structure. Up-regulation of telomerase immediate effect on urothelial carcinoma (UC) cell line expression occurs in the majority of urothelial carcinoma viability, before sufficient divisions to account for (UC) irrespective or stage or grade (1), suggesting that this telomere attrition, implicating non–telomere effects may be an early event in tumorigenesis. Normal human of telomerase in UC. We analyzed the effects of urothelial cells (NHUC) are immortalized by expression of telomerase on gene expression in isogenic mortal and hTERT, the catalytic subunit of telomerase. In contrast to hTERT-transduced NHUC. hTERT expression led to requirements for immortalization in other epithelial cell types consistent alterations in the expression of genes and despite the common loss of expression of p16 in UC, predicted to be of phenotypic significance in inactivation of the CDKN2A locus (encoding p16 and tumorigenesis. -

The Clinical Utility of Optical Genome Mapping for the Assessment of Genomic Aberrations in Acute Lymphoblastic Leukemia
cancers Article The Clinical Utility of Optical Genome Mapping for the Assessment of Genomic Aberrations in Acute Lymphoblastic Leukemia Jonathan Lukas Lühmann 1,† , Marie Stelter 1,†, Marie Wolter 1, Josephine Kater 1, Jana Lentes 1, Anke Katharina Bergmann 1, Maximilian Schieck 1 , Gudrun Göhring 1, Anja Möricke 2, Gunnar Cario 2, Markéta Žaliová 3 , Martin Schrappe 2, Brigitte Schlegelberger 1, Martin Stanulla 4 and Doris Steinemann 1,* 1 Department of Human Genetics, Hannover Medical School, 30625 Hannover, Germany; [email protected] (J.L.L.); [email protected] (M.S.); [email protected] (M.W.); [email protected] (J.K.); [email protected] (J.L.); [email protected] (A.K.B.); [email protected] (M.S.); [email protected] (G.G.); [email protected] (B.S.) 2 Department of Pediatrics I, ALL-BFM Study Group, Christian-Albrechts University Kiel and University Medical Center Schleswig-Holstein, 24105 Kiel, Germany; [email protected] (A.M.); [email protected] (G.C.); [email protected] (M.S.) 3 Department of Paediatric Haematology and Oncology, 2nd Faculty of Medicine, Charles University and University Hospital Motol, CZ-15006 Prague, Czech Republic; [email protected] 4 Pediatric Hematology and Oncology, Hannover Medical School, 30625 Hannover, Germany; [email protected] * Correspondence: [email protected] † These authors equally contributed to this work. Citation: Lühmann, J.L.; Stelter, M.; Wolter, M.; Kater, J.; Lentes, J.; Bergmann, A.K.; Schieck, M.; Simple Summary: The stratification of childhood ALL is currently based on various diagnostic Göhring, G.; Möricke, A.; Cario, G.; assays. -

Human Urinary Exosomes As Innate Immune Effectors
BASIC RESEARCH www.jasn.org Human Urinary Exosomes as Innate Immune Effectors † † ‡ Thomas F. Hiemstra,* Philip D. Charles, Tannia Gracia, Svenja S. Hester,§ † ‡ | Laurent Gatto, Rafia Al-Lamki,* R. Andres Floto,* Ya Su, Jeremy N. Skepper, † ‡ Kathryn S. Lilley, and Fiona E. Karet Frankl *Department of Medicine, †Cambridge Centre for Proteome Research and Cambridge Systems Biology Centre, Department of Biochemistry, ‡Department of Medical Genetics, and |Multi-Imaging Centre, Department of Anatomy, University of Cambridge, Cambridge, United Kingdom; and §Sir William Dunn School of Pathology, University of Oxford, Oxford, United Kingdom ABSTRACT Exosomes are small extracellular vesicles, approximately 50 nm in diameter, derived from the endocytic pathway and released by a variety of cell types. Recent data indicate a spectrum of exosomal functions, including RNA transfer, antigen presentation, modulation of apoptosis, and shedding of obsolete protein. Exosomes derived from all nephron segments are also present in human urine, where their function is unknown. Although one report suggested in vitro uptake of exosomes by renal cortical collecting duct cells, most studies of human urinary exosomes have focused on biomarker discovery rather than exosome function. Here, we report results from in-depth proteomic analyses and EM showing that normal human urinary exosomes are significantly enriched for innate immune proteins that include antimicrobial proteins and peptides and bacterial and viral receptors. Urinary exosomes, but not the prevalent soluble urinary protein uromodulin (Tamm–Horsfall protein), potently inhibited growth of pathogenic and commensal Escherichia coli and induced bacterial lysis. Bacterial killing depended on exosome structural integrity and occurred optimally at the acidic pH typical of urine from omnivorous humans. -

GOLGA5 Purified Maxpab Rabbit Polyclonal Antibody (D01P)
GOLGA5 purified MaxPab rabbit polyclonal antibody (D01P) Catalog # : H00009950-D01P 規格 : [ 100 ug ] List All Specification Application Image Product Rabbit polyclonal antibody raised against a full-length human GOLGA5 Western Blot (Tissue lysate) Description: protein. Immunogen: GOLGA5 (NP_005104.2, 1 a.a. ~ 731 a.a) full-length human protein. Sequence: MSWFVDLAGKAEDLLNRVDQGAATALSRKDNASNIYSKNTDYTELHQQN TDLIYQTGPKSTYISSAADNIRNQKATILAGTANVKVGSRTPVEASHPVEN enlarge ASVPRPSSHFVRRKKSEPDDELLFDFLNSSQKEPTGRVEIRKEKGKTPV FQSSQTSSVSSVNPSVTTIKTIEENSFGSQTHEAASNSDSSHEGQEESSK Western Blot (Tissue lysate) ENVSSNAACPDHTPTPNDDGKSHELSNLRLENQLLRNEVQSLNQEMASL LQRSKETQEELNKARARVEKWNADHSKSDRMTRGLRAQVDDLTEAVAA KDSQLAVLKVRLQEADQLLSTRTEALEALQSEKSRIMQDQSEGNSLQNQ ALQTFQERLHEADATLKREQESYKQMQSEFAARLNKVEMERQNLAEAIT LAERKYSDEKKRVDELQQQVKLYKLNLESSKQELIDYKQKATRILQSKEK LINSLKEGSGFEGLDSSTASSMELEELRHEKEMQREEIQKLMGQIHQLRS ELQDMEAQQVNEAESAREQLQDLHDQIAGQKASKQELETELERLKQEF HYIEEDLYRTKNTLQSRIKDRDEEIQKLRNQLTNKTLSNSSQSELENRLHQ enlarge LTETLIQKQTMLESLSTEKNSLVFQLERLEQQMNSASGSSSNGSSINMSG IDNGEGTRLRNVPVLFNDTETNLAGMYGKVRKAASSIDQFSIRLGIFLRRY Western Blot (Cell lysate) PIARVFVIIYMALLHLWVMIVLLTYTPEMHHDQPYGK Host: Rabbit Reactivity: Human, Mouse enlarge Quality Control Antibody reactive against mammalian transfected lysate. Testing: Western Blot (Transfected lysate) Storage Buffer: In 1x PBS, pH 7.4 Storage Store at -20°C or lower. Aliquot to avoid repeated freezing and thawing. Instruction: MSDS: Download enlarge Datasheet: Download Immunofluorescence Applications Western Blot (Tissue lysate) -
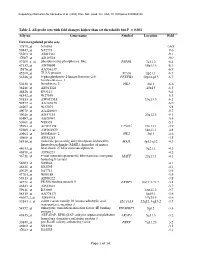
Table 2. All Probe Sets with Fold Changes Higher Than Set Thresholds but P > 0.001 Affy No
Supporting information for Sanoudou et al. (2003) Proc. Natl. Acad. Sci. USA, 10.1073/pnas.0330960100 Table 2. All probe sets with fold changes higher than set thresholds but P > 0.001 Affy no. Gene name Symbol Location Fold Down-regulated probe sets 47879_at N46863 -16.9 59447_at N52773 -9.6 55503_at AI085361 -9.1 47087_at AI310524 -9.0 37209_g_at phosphoserine phosphatase-like PSPHL 7q11.2 -8.4 47137_at AI479899 19p13.3 -8.1 45876_at AA536137 -6.9 45260_at TU3A protein TU3A 3p21.1 -6.7 56246_at 6-phosphofructo-2-kinase/fructose-2,6- PFKFB3 10p14-p15 -6.7 bisphosphatase 3 50230_at hexokinase 2 HK2 2p13 -6.6 38248_at AB011124 20p13 -6.5 44426_at R93141 -6.4 48542_at W27559 -6.3 53813_at AW051518 13q13.3 -6.1 59577_at AA243670 -6.0 46907_at W37075 -5.8 49078_at AA424983 -5.7 49026_at AI357153 20p12.3 -5.4 53487_at AI670947 -5.4 50161_at N39328 -5.1 35994_at AC002398 F25965 19q13.1 -4.9 52285_f_at AW002970 18p11.1 -4.8 40964_at hexokinase 2 HK2 2p13 -4.6 49806_at AI932283 -4.5 58918_at molecule possessing ankyrin repeats induced by MAIL 3p12-q12 -4.3 lipopolysaccharide (MAIL), homolog of mouse 44633_at heat shock 27 kDa associated protein 3q21.1 -4.3 46858_at AI796221 -4.2 36711_at v-maf musculoaponeurotic fibrosarcoma oncogene MAFF 22q13.1 -4.1 homolog F (avian) 54683_at N49844 -4.1 46621_at N32595 -4.1 49629_at N47713 -3.9 47703_at W89189 -3.9 59313_at AI598222 -3.8 34721_at FK506 binding protein 5 FKBP5 6p21.3-21.2 -3.8 46843_at AI632621 -3.7 59611_at R53069 16p11.2 -3.7 58315_at AA778171 3p25.1 -3.6 46607_f_at AI885018 17q25.3 -3.6 33143_s_at solute carrier family 16 (monocarboxylic acid SLC16A3 22q12.3-q13.2 -3.5 transporters), member 3 54152_at eukaryotic translation initiation factor 4E binding EIF4EBP1 8p12 -3.4 protein 1 43935_at ARF-GAP, RHO-GAP, ankyrin repeat and plekstrin ARAP3 5q31.3 -3.2 homology domains-containing protein 3 33849_at pre-B-cell colony-enhancing factor PBEF 7q11.23 -3.2 46902_at N92294 -3.2 47023_at N25555 -3.1 Page 1 of 14 Supporting information for Sanoudou et al. -

The Regulation of Self-Renewal in Normal Human Urothelial Cells
The Regulation of Self-Renewal in Normal Human Urothelial Cells Lisa A. Kirkwood PhD University of York Department of Biology April 2012 Abstract The urinary tract is lined by a mitotically-quiescent, but highly regenerative epithelium, the urothelium. The mechanisms regulating urothelial regeneration are incompletely understood although autocrine stimulation of the Epidermal Growth Factor Receptor (EGFR) signalling pathway has been implicated. The hypothesis developed in this thesis is that urothelial homeostasis is regulated through resolution of interactive signal transduction networks downstream of local environmental cues, such as cell:cell contact. Here, canonical Wnt signalling was examined as a candidate key pathway due to the pivotal role of β-catenin in both nuclear transcription and intercellular adherens junctions. Normal human urothelial (NHU) cells isolated from surgical biopsies were grown as finite cell lines in monolayer culture. mRNA analysis from proliferating cultures inferred all components for a functional autocrine-activated canonical Wnt cascade were present. In proliferating cells, β-catenin was nuclear and Axin2 expression provided an objective hallmark of β-catenin/TCF transcription factor activity. This endogenous activity was not mediated by Wnt receptor activation, as Wnt ligand was produced in inactive (non-palmitylated) form in serum-free culture, but instead -catenin activation was driven via EGFR- mediated phosphorylation of GSK3 and inhibition of the β-catenin destruction complex. In quiescent, contact–inhibited cultures, β-catenin was seen to re- localise to the adherens junctions and GSK3β activity was re-established. Knock-down of β-catenin using RNA interference led to significant changes in p-ERK and p-AKT activity as well as an increase in E-cadherin protein expression. -

1 Novel Expression Signatures Identified by Transcriptional Analysis
ARD Online First, published on October 7, 2009 as 10.1136/ard.2009.108043 Ann Rheum Dis: first published as 10.1136/ard.2009.108043 on 7 October 2009. Downloaded from Novel expression signatures identified by transcriptional analysis of separated leukocyte subsets in SLE and vasculitis 1Paul A Lyons, 1Eoin F McKinney, 1Tim F Rayner, 1Alexander Hatton, 1Hayley B Woffendin, 1Maria Koukoulaki, 2Thomas C Freeman, 1David RW Jayne, 1Afzal N Chaudhry, and 1Kenneth GC Smith. 1Cambridge Institute for Medical Research and Department of Medicine, Addenbrooke’s Hospital, Hills Road, Cambridge, CB2 0XY, UK 2Roslin Institute, University of Edinburgh, Roslin, Midlothian, EH25 9PS, UK Correspondence should be addressed to Dr Paul Lyons or Prof Kenneth Smith, Department of Medicine, Cambridge Institute for Medical Research, Addenbrooke’s Hospital, Hills Road, Cambridge, CB2 0XY, UK. Telephone: +44 1223 762642, Fax: +44 1223 762640, E-mail: [email protected] or [email protected] Key words: Gene expression, autoimmune disease, SLE, vasculitis Word count: 2,906 The Corresponding Author has the right to grant on behalf of all authors and does grant on behalf of all authors, an exclusive licence (or non-exclusive for government employees) on a worldwide basis to the BMJ Publishing Group Ltd and its Licensees to permit this article (if accepted) to be published in Annals of the Rheumatic Diseases and any other BMJPGL products to exploit all subsidiary rights, as set out in their licence (http://ard.bmj.com/ifora/licence.pdf). http://ard.bmj.com/ on September 29, 2021 by guest. Protected copyright. 1 Copyright Article author (or their employer) 2009.