3.1 Polarization Behavior of Active Passive Metals and Alloys Glossary
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Absolute Measurement of the Abundance of Atmospheric Carbon Monoxide C
Absolute measurement of the abundance of atmospheric carbon monoxide C. Brenninkmeijer, C. Koeppel, T. Röckmann, D. Scharffe, Maya Bräunlich, Valerie Gros To cite this version: C. Brenninkmeijer, C. Koeppel, T. Röckmann, D. Scharffe, Maya Bräunlich, et al.. Absolute mea- surement of the abundance of atmospheric carbon monoxide. Journal of Geophysical Research: At- mospheres, American Geophysical Union, 2001, 106 (D9), pp.10003-10010. 10.1029/2000JD900342. hal-03117189 HAL Id: hal-03117189 https://hal.archives-ouvertes.fr/hal-03117189 Submitted on 8 Feb 2021 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. JOURNAL OF GEOPHYSICAL RESEARCH, VOL. 106, NO. D9, PAGES 10,003-10,010, MAY 16, 2001 Absolute measurement of the abundance of atmospheric carbon monoxide C. A.M. Brenninkmeijer,C. Koeppel,T. R6ckmann,D. S. Scharffe, Maya Br•iunlich,and Valerie Gros AtmosphericChemistry Division, Max PlanckInstitute for Chemistry,Mainz, Germany Abstract.The main aspects of anabsolute method for measurementof the mixing ratio of atmos- phericcarbon monoxide (CO) are presented. The method is based on cryogenic extraction of CO fromair afteroxidation to CO2followed by accuratevolumetric determination. Gravimetric meas- urementis usedto determinethe quantity of sampleair processed.In routineoperation the overall errorcan be kept below 1%. -

Effects on Accuracy and Detection Limits
WHITE Sensitivity, Background, Noise, and Calibration in Atomic PAPER Spectroscopy: Effects on Accuracy and Detection Limits Introduction Proper calibration in atomic spectroscopy and an understanding The three most common atomic spectroscopy techniques of uncertainty is fundamental to achieving accurate results. This are atomic absorption (AA) spectroscopy, ICP optical emission paper provides a practical discussion of the effects of noise, error spectroscopy (ICP-OES) and ICP mass spectrometry (ICP-MS). and concentration range of calibration curves on the ability to Of these, ICP-OES and ICP-MS are very linear; that is, a plot determine the concentration of a given element with reasonable of concentration vs. intensity forms a straight line over a accuracy. The determination of lower limits of quantitation and wide range of concentrations (Figure 1). AA is linear over a lower limits of detection will also be discussed. much smaller range and begins to curve downward at higher Results accuracy is highly dependent on blank contamination, concentrations (Figure 2). Linear ranges are well understood, linearity of calibration standards, curve-fitting choices and the and, for AA, a rule of thumb can be applied to estimate the range of concentrations chosen for calibration. Additional factors maximum working range using a non-linear algorithm. include the use of internal standards (and proper selection of This paper will discuss the contributions of sensitivity, internal standards) and instrumental settings. background, noise, calibration range, calibration mathematics This paper is not intended to be a rigorous treatment of statistics and contamination on the ability to achieve best accuracy and in calibration; many references are available for this, such as lowest detection limits. -

Combined Wastestream Formula (CWF)
UnitedStates PermitsDivision and September1985 EnvironmentalProtection IndustrialTechnology Division Agency Washington,DC 20460 Water EPA Guidance Manual Pretreatment for the Use of Production-Based Pretreatment Standards and the Combined Wastestream Formula UNITED STATES ENVIRONMENTAL PROTECTION AGENCY WASHINGTON, D.C. 20460 SEP 19 1985 OFFICEOF WATER MEMORANDUM SUBJECT: Pretreatment Program Guidance FROM: Rebecca W. Hanmer, Director Office of Water Enforcement and Permits (EN-335) James M. Conlon, Acting Director Office of Water Regulations and Standards (WH-551) TO: Users of the Guidance Manual for the Use of Production- Based Categorical Pretreatment Standards and the Combined Wastestream Formula This guidance manual has been developed by EPA to explain how to implement two important elements of the national pretreatment program: categorical standards and the combined wastestream formula. The manual is divided into two sections. The first section explains how to apply production-based categorical standards in a permit, contract, or similar mechanism. The second part explains how to use the combined wastestream formula, providing definitions and examples. The manual is one of a series of guidance documents intended to simplify and improve understanding of various aspects of the pretreatment program. Other documents in this series which have either been recently issued or will be issued in the near future will provide guidance on: 1) Removal Credits 2) Total Toxic Organics (TTO) Monitoring 3) RCRA Notification Requirements 4) Local Limits 5) POTW Interference The need for guidance on the use of categorical standards and the combined wastestream formula was recognized by the Pretreatment Implementation Review Task Force (PIRT). PIRT was set up by the EPA Administrator to make recommendations concerning the problems faced by POTWs, states, and industry in implementing the national pretreatment program. -

Florpyrauxifen-Benzyl & Degradates in Compost
Page 16 INTRODUCTION Scope The objective of the study was to independently validate analytical method as given in the DAS study 171407 [1] for the determination of Florpyrauxifen benzyl (XDE-848 BE), X11438848 and X11966341 in compost in accordance to the guidance documents SANCO/825/00, rev. 8.1 [2] and SANCO/3029/99 rev. 4 of the European Commission [3], and OCSPP 850.6100 of the United States Environmental Protection Agency [4]. The limit of quantification was 0.00015 mg/kg for Florpyrauxifen benzyl (XDE-848 BE), 0.00045 mg/kg for X11438848 and 0.009 mg/kg for X11966341 Analytical Procedure Compost samples were extracted by shaking with acetonitrile/0.1N hydrochloric acid (90:10, v/v), centrifuging, and decanting into a separate tube containing QuEChERS Citrat kit. An aliquot of 1N HCl was added to the extract followed by centrifugation. After a portion of the organic layer was aliquoted, internal standard was added and the sample was evaporated to near dryness under a stream of nitrogen. 1N HCl was added and the samples were incubated for one hour at 80 °C. Ethyl acetate was added and the samples were transferred to a Supel QuE Z-Sep tube and centrifuged. After centrifuging, the samples were placed in a dry ice bath to flash freeze the aqueous layer and the organic layer was poured off into a glass tube. The samples were evaporated under a stream of nitrogen, reconstituted in methanol and 0.1% formic acid in water, and transferred into an autosampler vial for analysis. The samples were analyzed for XDE-848 BE and its metabolites by liquid chromatography with positive ion electrospray ionization tandem mass spectrometry Selectivity Quantification was performed by use of LC-MS/MS detection. -

Solution Density Modelling for Single and Mixed Base Metal Electrolytes at Ionic Level
CORE Metadata, citation and similar papers at core.ac.uk Provided by Wits Institutional Repository on DSPACE SOLUTION DENSITY MODELLING FOR SINGLE AND MIXED BASE METAL ELECTROLYTES AT IONIC LEVEL Trevor Chagonda A research report submitted to the Faculty of Engineering and the Built Environment, University of the Witwatersrand, in partial fulfilment of the requirements for the degree of Master of Science in Metallurgical Engineering Johannesburg 2013 DECLARATION I declare that this research report is my own unaided work. It is being submitted in partial fulfilment of the requirements of the degree of Master of Science in Metallurgical Engineering at the University of Witwatersrand, Johannesburg. It has not been submitted before for any degree or examination to any other university. ……………………………………. (Signature of Candidate) …………….. DAY OF ……………………………. YEAR …………………… ABSTRACT Solution density modeling is important in hydrometallurgical processes as accurate predictions of single and mixed electrolytes can be used in the design of equipment and their sizing, heat transfer calculations and choosing of materials for construction. This research project entails modeling of electrolyte solutions by extending the Laliberte and Cooper (compound level) model to ionic level where an electrolyte solution is modeled as a mixture of cations, anions and water molecules. This modeling predicts single and mixed electrolyte density as a function of electrolyte temperature in degrees Celsius; water, cation and anion apparent volumes in cubic centimeters; and their respective concentrations in the electrolyte as mass fractions. The model was developed by fitting single electrolyte density data reported in literature using the least squares method in Microsoft Excel®. The following 26 single electrolyte solutions were used in the fitting exercise: Al2(SO4)3, BaCl2, CaCl2, CdSO4, CoCl2, CuSO4, FeCl3, FeSO4, HCl, HCN, HNO3, K2CO3, LiCl, MgSO4, MnCl2, Na2SO3, NaF, NaI, NaOH, (NH4)2SO4, NiCl2, SrCl2, ZnCl2, ZnBr2, (NH4)2C2O4 and KNO2. -

Human Health Criteria Options February 2017
Water Quality Policy 1-11 Updates Human Health Criteria February 2017 Outline of Issues covered: I. Background II. Factors to consider for the use of tissue samples Tissue sample representativeness Species to represent segment/site fidelity III. Factors to consider for the use of water samples IV. Lines of Evidence Tissue exposure concentration Drinking water exposure concentration Water column data DOH fish advisories Safe Drinking Water Act (SDWA) MCL values V. Weight of evidence VI. Category determinations I. Background The surface water quality standards include a section on toxic substances (WAC 173-201A-240) for the protection of designated uses. Numeric criteria have been developed for the designated uses of aquatic life and the protection of human health. Uses associated with human health are fish and shellfish harvesting (consumption of fish) and domestic water supply (drinking water). Human health-based water quality criteria used by the state prior to December 2016 were contained in the National Toxics Rule. Currently approved human health criteria (HHC) for Washington are a combination of state-adopted criteria and EPA promulgated criteria. In previous Water Quality Assessments, toxics data from fish tissue was used to assess for the protection of human health, deriving a Fish Tissue Equivalent Concentration (FTEC) as a means to back-calculate fish tissue pollutant concentrations to approximate surface water pollutant concentrations. This was done by applying bioconcentration factors (BCF) that were used to derive the HHC in the National Toxics Rule. The intent of the FTEC used for the past several years was to represent a water column criteria equivalent. On December 14, 2016, Ecology held a public dialogue session on human health criteria. -

Calculation of the Electrophoretic Mobility of a Spherical Colloid Particle
JOUI~NziL OF COLLOID £ND INTERFACE SCIENCE 9.2, 78--99 (1966) Calculation of the Electrophoretic Mobility of a Spherical Colloid Particle P. H. WIERSEMA, A. L. LOEB, AXD J. TH. G. OVERBEEK Van't Hoff Laboratory, University of Utrecht, The Netherlands. Ledgemont Laboratory, Kennecott Copper Corporation, Lexington, Massachusetts Received May 3, 1965 ABSTRACT A new calculation of the relation between the electrophoretic mobility and the/-- potential of a spherical colloid particle is presented. The model consists of a rigid, electrically insulating sphere surrounded by a Gouy-Chapman double layer. The ap- propriate differential equations (which account for both electrophoretic retardation and relaxation effect) have been solved without approximations on an IBM 704 com- puter. The theoretical assumptions and the basic equations ~re stated. A detailed account of the solutions of the equations is pub]ished elsewhere. The present paper contains the complete results. Comparison of these results with those of Overbeek ~nd of Booth shows that, %r high ~--potential ~nd 0.2 < ~a < 50, their approximations gen- erally overestimate the relaxation effect. The application to practical cases (espe- cially colloid particles in solutions of 1-1 electrolytes) is discussed extensively. Finally, the theoretical results are compared with experimental data published by others. INTRODUCTION particle, it is valid only when ~a }> 1, The calculation of the ~-potential from where a is the radius of the particle and measurements of the electrophoretic mobil- is the reciprocal thickness of the surround- ity (E.M.) requires a theoretical relation ing ionic atmosphere. For moderate values between the two quantities. The oldest of ~a (say, 0.2 < ~a < 50), the so-called relation of this kind, which was derived by relaxation effect leads to important correc- yon Helmholtz (1) and improved by yon tions, which increase with increasing /'- Smoluchowski (2), reads potential. -

Test Chemical Solubility Protocol
Test Method Protocol for Solubility Determination Phase III - Validation Study September 24, 2003 TEST METHOD PROTOCOL for Solubility Determination In Vitro Cytotoxicity Validation Study Phase III September 24, 2003 Prepared by The National Toxicology Program (NTP) Interagency Center for the Evaluation of Alternative Toxicological Methods (NICEATM) Based on Standard Operating Procedure Recommendations from an International Workshop Organized by the Interagency Coordinating Committee on the Validation of Alternative Methods (ICCVAM) National Institute of Environmental Health Sciences (NIEHS) National Institutes of Health (NIH) U.S. Public Health Service Department of Health and Human Services 1 Test Method Protocol for Solubility Determination Phase III - Validation Study September 24, 2003 TEST METHOD PROTOCOL Solubility Determination Phase III I. PURPOSE The purpose of this study is to evaluate the cytotoxicity of test chemicals using the BALB/c 3T3 Neutral Red Uptake (NRU) and normal human keratinocyte (NHK) cytotoxicity tests. The data will be used to evaluate the intra- and inter-laboratory reproducibility of the assay and effectiveness of the cytotoxicity assay to predict the starting doses for rodent acute oral systemic toxicity assays. This test method protocol outlines the procedures for performing solubility determinations for the in vitro validation study organized by NICEATM and the European Centre for the Validation of Alternative Methods (ECVAM) and sponsored by NIEHS, U.S. Environmental Protection Agency, and ECVAM. This test method protocol applies to all personnel involved with performing the solubility testing. A. Solubility Test The solubility tests will be performed to determine the best solvent to use for each of the 60 blinded/coded test chemicals to be tested in the 3T3 and NHK NRU cytotoxicity tests II. -

Calculating Water Quality-Based Effluent Limitations for Surface Water Discharges
BUREAU OF WATER QUALITY PROGRAM GUIDANCE Wastewater Policy and Management Team Wisconsin Department of Natural Resources 101 S. Webster Street, P.O. Box 7921 Madison, WI 53707-7921 Calculating Water Quality-Based Effluent Limitations for Surface Water Discharges 08/10/2020 EGAD Number: 3400-2020-13 This document is intended solely as guidance and does not contain any mandatory requirements except where requirements found in statute or administrative rule are referenced. Any regulatory decisions made by the Department of Natural Resources in any matter addressed by this guidance will be made by applying the governing statutes and administrative rules to the relevant facts. _____________________________________________________________________________________ APPROVED: Adrian Stocks 12/07/2020 _______________________________ __________ Adrian Stocks, Director Date Bureau of Water Quality Summary Section 283.13(5), Wis. Stats., requires that effluent limitations be established in permits for point source discharges to surface water to ensure that applicable state water quality standards are met. These types of effluent limitations are protective of water quality and are referred to as water quality- based effluent limits (WQBEL). The WQBEL calculation process completed by the Wisconsin Department of Natural Resources (the department) involves a site-specific evaluation of the discharge receiving water and effluent characteristics and comparison to state water quality standards. This guidance is intended for use by department staff who are responsible for calculating WQBELs. The document covers how WQBELs are calculated, how staff determine if limits are needed in permits, and how those limits should be expressed in WPDES permits for point source discharges to Wisconsin surface waters. The following guidance documents also provide information on WQBEL calculation procedures and are used by WQBEL calculation staff. -

Sociation Constant of the Buffer and the Concentration and Reaction of the Buffer Solution.*
ON THE MEASUREMENT OF BUFFER VALUES AND ON THE RELATIONSHIP OF BUFFER VALUE TO THE DIS- SOCIATION CONSTANT OF THE BUFFER AND THE CONCENTRATION AND REACTION OF THE BUFFER SOLUTION.* BY DONALD D. VAN SLYKE. (From the Hospital of The Rockefeller Institute for Medical Research.) Downloaded from (Received for publication, April 15, 1922.) CONTENTS. The nature and mode of action of buffers ............................ 525 Unit for measurement of buffer values ............................... 528 The buffer value of water plus only strong acid or alkali ............. 531 www.jbc.org The buffer value of a solution containing a weak monovalent acid anditssalt .................................................... 535 Fundamental mass law equations ................................ 535 Differentiation of Henderson’s equation in order to calculate the buffer values of weak acids .............................. 537 by guest, on September 19, 2010 Second derivative of Henderson’s equation. Point of maxi- mum buffer value ........................................... 542 General equation indicating the dissociation of a weak acid at all reactions within the limits of validity of the mass law ... 547 Polyvalent acid buffers and mixtures of monovalent acid buffers ..... 553 Basicbuffers ........................................................ 557 Amphotericbuffers .................................................. 558 Universalbuffermixtures ............................................. 558 The buffer value of blood ........................................... -

Corrosion and Deuterium Uptake of Zr-Based Alloys in Supercritical Water
CW-124680-CONF-001 UNRESTRICTED - ILLIMITÉ Corrosion and Deuterium Uptake of Zr-based Alloys In Supercritical Water D. Khatamian AECL, Chalk River Laboratories, Stn. 55 Chalk River, ON K0J 1J0, Canada [email protected] Abstract To increase the thermodynamic efficiency above 40% in nuclear power plants, the use of supercritical water as the heat transport fluid has been suggested. Zircaloy-2, -4, Zr-Cr-Fe, Zr-1Nb and Zr-2.5Nb were tested as prospective fuel cladding materials in 30 MPa D2O at 500 C. Zircaloy-2 showed the highest rates of corrosion and hydriding. Although Zr-Cr-Fe initially showed a very low corrosion rate, it displayed breakaway corrosion kinetics after 50 h exposure. The best-behaved material both from a corrosion and hydrogen uptake point of view was Zr-2.5Nb. However, the Zr-2.5Nb oxide growth rate was still excessive and beyond the current CANDU design allowance. Similar coupons, coated with Cr, were also tested. The coated layer effectively prevented oxidation of the coupons except on the edges, where the coating was thinner and had some flaws. In addition, the Cr-coated Zr-2.5Nb coupons had the lowest deuterium pickup of all the alloys tested and showed no signs of accelerated or non- uniform corrosion. CANDU–CANada Deuterium Uranium is a registered trademark of Atomic Energy of Canada Ltd. 2 CW-124680-CONF-001 UNRESTRICTED - ILLIMITÉ 1.0 INTRODUCTION In a nuclear power plant, increasing thermodynamic efficiency and simplifying plant design are the two major ways of reducing unit energy cost. To increase the thermodynamic efficiency above 40%, the use of high-pressure supercritical water at 500 C and above, as the heat transport fluid (HTF), has been suggested [1,2]. -
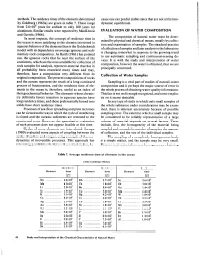
Methods. the Residence Times of the Elements Determined by Goldberg
methods.The residencetimes of the elementsdetermined causeone can predict stable statesthat are not at thermo- by Goldberg (1963a) are given in table 7. These range dynamic equilibrium. from 2.6~10~ years for sodium to only 100 years for aluminum. Similar results were reported by MacKenzie EVALUATION OF WATER COMPOSITION and Garrels (1966). The composition of natural water must be deter- In most respects, the concept of residence time in mined by physical and chemical means,usually by collec- the ocean is more satisfying to the chemist interested in tion and examination of samples.The standard practice aqueousbehavior of the elementsthan is the Goldschmidt of collection of samplesand later analysisin the laboratory model with its dependenceon averageigneous and sedi- is changing somewhat in responseto the growing trend mentary rock composition. As Barth (1961) has pointed to use automatic sampling and continuous-sensing de- out, the igneous rocks that lie near the surface of the vices. It is with the study and interpretation of water continents, which are the ones available for collection of composition, however the water is obtained, that we are rock samples for analysis, represent material that has in principally concerned. all probability been reworked many times and may, therefore, have a composition very different from its Collection of Water Samples original composition. The present compositions of rocks and the oceans represent the result of a long-continued Sampling is a vital part of studies of natural-water process of fractionation, and the residence time of ele- composition and is perhaps the major source of error in ments in the ocean is, therefore, useful as an index of the whole processof obtaining water-quality information.