Characterization of Fraxinus Spp. Phloem Transcriptome Thesis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Dissert 1 Title
GENETIC AND BIOCHEMICAL ANALYSIS OF ESSENTIAL ENZYMES IN TRIACYLGLYCEROL SYNTHESIS IN ARABIDOPSIS By KIMBERLY LYNN COTTON A dissertation submitted in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY WASHINGTON STATE UNIVERSITY Molecular Plant Sciences DECEMBER 2015 © Copyright by KIMBERLY LYNN COTTON, 2015 All Rights Reserved To the Faculty of Washington State University: The members of the Committee appointed to examine the dissertation of KIMBERLY LYNN COTTON find it satisfactory and recommend that it be accepted. ____________________________________________ John A. Browse, Ph.D., Chair _____________________________________________ Michael M. Neff, Ph.D. _____________________________________________ Thomas W. Okita, Ph.D. _____________________________________________ John J. Wyrick, Ph.D. ii ACKNOWLEDGMENTS To my PI John Browse, thank you for your support, guidance and patience over the years. To my committee, John Browse, Michael Neff, Thomas Okita, and John Wyrick, thank you for your insightful discussions and suggestions. To my collaborators Jay Shockey, Anushobha Regmi, Neil Adhikari, John Browse, and Phil Bates, thanks for the great work and great times. To Browse Lab members past and present, thanks for the help, guidance and wonderful memories. To the undergraduate students who have helped me over the years, thank you for all your help in making this project possible; and with a special thank you to Arlene, Tesa, Breana, and “no-longer-undergrads” David and Barbara Cotton for their particular dedication to this project. And finally, a huge thank you to my family and friends who have loved and supported me throughout this venture. Go Cougs! iii GENETIC AND BIOCHEMICAL ANALYSIS OF ESSENTIAL ENZYMES IN TRIACYLGLYCEROL SYNTHESIS IN ARABIDOPSIS Abstract by Kimberly Lynn Cotton, Ph.D. -

Transcriptional Regulation of RKIP in Prostate Cancer Progression
Health Science Campus FINAL APPROVAL OF DISSERTATION Doctor of Philosophy in Biomedical Sciences Transcriptional Regulation of RKIP in Prostate Cancer Progression Submitted by: Sandra Marie Beach In partial fulfillment of the requirements for the degree of Doctor of Philosophy in Biomedical Sciences Examination Committee Major Advisor: Kam Yeung, Ph.D. Academic William Maltese, Ph.D. Advisory Committee: Sonia Najjar, Ph.D. Han-Fei Ding, M.D., Ph.D. Manohar Ratnam, Ph.D. Senior Associate Dean College of Graduate Studies Michael S. Bisesi, Ph.D. Date of Defense: May 16, 2007 Transcriptional Regulation of RKIP in Prostate Cancer Progression Sandra Beach University of Toledo ACKNOWLDEGMENTS I thank my major advisor, Dr. Kam Yeung, for the opportunity to pursue my degree in his laboratory. I am also indebted to my advisory committee members past and present, Drs. Sonia Najjar, Han-Fei Ding, Manohar Ratnam, James Trempe, and Douglas Pittman for generously and judiciously guiding my studies and sharing reagents and equipment. I owe extended thanks to Dr. William Maltese as a committee member and chairman of my department for supporting my degree progress. The entire Department of Biochemistry and Cancer Biology has been most kind and helpful to me. Drs. Roy Collaco and Hong-Juan Cui have shared their excellent technical and practical advice with me throughout my studies. I thank members of the Yeung laboratory, Dr. Sungdae Park, Hui Hui Tang, Miranda Yeung for their support and collegiality. The data mining studies herein would not have been possible without the helpful advice of Dr. Robert Trumbly. I am also grateful for the exceptional assistance and shared microarray data of Dr. -

Downloaded in August 2017; 209326 Sequences)
microorganisms Article Label-Free Quantitative Proteomics Analysis in Susceptible and Resistant Brassica napus Cultivars Infected with Xanthomonas campestris pv. campestris Md Tabibul Islam 1,2, Bok-Rye Lee 1,3 , Van Hien La 1, Dong-Won Bae 4, Woo-Jin Jung 5 and Tae-Hwan Kim 1,* 1 Department of Animal Science, Institute of Agricultural Science and Technology, College of Agriculture & Life Science, Chonnam National University, Gwangju 61186, Korea; [email protected] (M.T.I.); [email protected] (B.-R.L.); [email protected] (V.H.L.) 2 Alson H. Smith Jr. Agricultural Research and Extension Center, School of Plant and Environmental Sciences, Virginia Tech, Winchester, VA 22602, USA 3 Asian Pear Research Institute, Chonnam National University, Gwangju 61186, Korea 4 Central Instrument Facility, Gyeongsang National University, Jinju 52828, Korea; [email protected] 5 Division of Applied Bioscience and Biotechnology, Institute of Environmentally Friendly Agriculture (IEFA), College of Agriculture and Life Science, Chonnam National University, Gwangju 61186, Korea; [email protected] * Correspondence: [email protected]; Tel.: +82-62-530-2126 Abstract: Black rot, caused by Xanthomonas campestris pv. campestris (Xcc), is the main disease of cruciferous vegetables. To characterize the resistance mechanism in the Brassica napus–Xcc pathosys- tem, Xcc-responsive proteins in susceptible (cv. Mosa) and resistant (cv. Capitol) cultivars were investigated using gel-free quantitative proteomics and analysis of gene expression. This allowed us to identify 158 and 163 differentially expressed proteins following Xcc infection in cv. Mosa and cv. Citation: Islam, M.T.; Lee, B.-R.; Capitol, respectively, and to classify them into five major categories including antioxidative systems, La, V.H.; Bae, D.-W.; Jung, W.-J.; proteolysis, photosynthesis, redox, and innate immunity. -

PANNZER 2: Annotate a Complete Proteome in Minutes!
PANNZER 2: Annotate a complete proteome in minutes! Alan J Medlar#, Petri Törönen#, Elaine Zosa, Liisa Holm* Structural Genomics Group, Institute of Biotechnology PO Box 56, 00014 University of Helsinki, Finland #These authors contributed equally *To whom correspondence should be addressed: [email protected] 1. INTRODUCTION As high-throughput sequencing has become increasingly efficient, downstream analysis has become the main bottleneck in genome sequencing projects. For example, a critical and time-consuming step is the annotation of the organism’s proteome: assigning molecular functions, involvement in biological processes and the cellular localizations of all identified protein sequences. The process of protein annotation involves classifying each protein using the Gene Ontology (GO) and selecting suitable free text descriptions, which are required for submission into sequence databases. We refer to these as GO and DE predictions, respectively. Although servers are available that perform functional annotation, none perform both GO and DE predictions concurrently, they tend to be slow and are restricted in the number of queries that can be submitted. Finally, many of the existing methods do not return an estimate of the prediction accuracy. 2. METHODS AND RESULTS The above shortcomings are remedied by PANNZER 2, an interactive web server and standalone program for protein function prediction. It is, to our knowledge, the only tool that performs both DE and GO prediction. It uses SANS-parallel [1], a high-throughput sequence search program that is thousands of times faster than BLAST, to identify homologous sequences. It is, therefore, capable of analyzing tens of thousands of proteins interactively. Results are displayed as an HTML table summarizing both DE and GO predictions, including a statistical estimate for the reliability of each prediction. -

Construction of Prognostic Microrna Signature for Human Invasive Breast Cancer by Integrated Analysis
Journal name: OncoTargets and Therapy Article Designation: Original Research Year: 2019 Volume: 12 OncoTargets and Therapy Dovepress Running head verso: Shi et al Running head recto: Shi et al open access to scientific and medical research DOI: 189265 Open Access Full Text Article ORIGINAL RESEARCH Construction of prognostic microRNA signature for human invasive breast cancer by integrated analysis This article was published in the following Dove Medical Press journal: OncoTargets and Therapy Wei Shi* Background: Despite the advances in early detection and treatment methods, breast cancer Fang Dong* still has a high mortality rate, even in those patients predicted to have a good prognosis. The Yujia Jiang purpose of this study is to identify a microRNA signature that could better predict prognosis in Linlin Lu breast cancer and add new insights to the current classification criteria. Changwen Wang Materials and methods: We downloaded microRNA sequencing data along with correspond- Jie Tan ing clinicopathological data from The Cancer Genome Atlas (TCGA). Of 1,098 breast cancer patients identified, 253 patients with fully characterized microRNA profiles were selected for Wen Yang analysis. A three-microRNA signature was generated in the training set. Subsequently, the per- Hui Guo formance of the signature was confirmed in a validation set. After construction of the signature, Jie Ming* we conducted additional experiments, including flow cytometry and the Cell Counting Kit-8 Tao Huang* assay, to illustrate the correlation of this microRNA signature with breast cancer cell cycle, Department of Breast and Thyroid apoptosis, and proliferation. Surgery, Union Hospital, Tongji Results: Three microRNAs (hsa-mir-31, hsa-mir-16-2, and hsa-mir-484) were identified to Medical College, Huazhong University of Science and Technology, Wuhan be significantly and independently correlated with patient prognosis, and performed with good 430022, China stability. -

Anti-Inflammatory Role of Curcumin in LPS Treated A549 Cells at Global Proteome Level and on Mycobacterial Infection
Anti-inflammatory Role of Curcumin in LPS Treated A549 cells at Global Proteome level and on Mycobacterial infection. Suchita Singh1,+, Rakesh Arya2,3,+, Rhishikesh R Bargaje1, Mrinal Kumar Das2,4, Subia Akram2, Hossain Md. Faruquee2,5, Rajendra Kumar Behera3, Ranjan Kumar Nanda2,*, Anurag Agrawal1 1Center of Excellence for Translational Research in Asthma and Lung Disease, CSIR- Institute of Genomics and Integrative Biology, New Delhi, 110025, India. 2Translational Health Group, International Centre for Genetic Engineering and Biotechnology, New Delhi, 110067, India. 3School of Life Sciences, Sambalpur University, Jyoti Vihar, Sambalpur, Orissa, 768019, India. 4Department of Respiratory Sciences, #211, Maurice Shock Building, University of Leicester, LE1 9HN 5Department of Biotechnology and Genetic Engineering, Islamic University, Kushtia- 7003, Bangladesh. +Contributed equally for this work. S-1 70 G1 S 60 G2/M 50 40 30 % of cells 20 10 0 CURI LPSI LPSCUR Figure S1: Effect of curcumin and/or LPS treatment on A549 cell viability A549 cells were treated with curcumin (10 µM) and/or LPS or 1 µg/ml for the indicated times and after fixation were stained with propidium iodide and Annexin V-FITC. The DNA contents were determined by flow cytometry to calculate percentage of cells present in each phase of the cell cycle (G1, S and G2/M) using Flowing analysis software. S-2 Figure S2: Total proteins identified in all the three experiments and their distribution betwee curcumin and/or LPS treated conditions. The proteins showing differential expressions (log2 fold change≥2) in these experiments were presented in the venn diagram and certain number of proteins are common in all three experiments. -
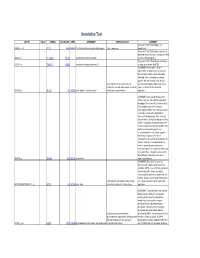
Supplementary Table 5. Functional Annotation of the Largest Gene Cluster(221 Element)
Annotation Tool AFFYID VALUE SYMBOL LOCUSLINK OMIM GENENAME GENEONTOLOGY SUMMARY [Proteome FUNCTION:] Expressed 203054_s_at TCTA 6988 600690 T-cell leukemia translocation altered gene tumor suppressor ubiquitously [Proteome FUNCTION:] May be involved in protein-protein interactions; contains five WD 44563_at FLJ10385 55135 hypothetical protein FLJ10385 domains (WD-40 repeats) [Proteome FUNCTION:] Weakly similarity to 212261_at TNRC15 26058 trinucleotide repeat containing 15 a region of rat nestin (Rn.9701) [SUMMARY:] Actin alpha 1 which is expressed in skeletal muscle is one of six different actin isoforms which have been identified. Actins are highly conserved proteins that are involved in cell motility, actin filament; motor activity; muscle structure and integrity. Alpha actins are a contraction; muscle development; structural major constituent of the contractile 203872_at ACTA1 58 102610 actin, alpha 1, skeletal muscle constituent of cytoskeleton apparatus. [SUMMARY:] Annexin VIII belong to the family of Ca (2+) dependent phospholipid binding proteins (annexins), and has a high 56% identity to annexin V (vascular anticoagulant-alpha). It was initially isolated as 2.2 kb vascular anticoagulant-beta transcript from human placenta, a Ca (2+) dependent phospholipid binding protein that inhibits coagulation and phospholipase A2 activity. However, the fact that annexin VIII is neither an extracellular protein nor associated with the cell surface suggests that it may not play a role in blood coagulation in vivo and its physiological role remains unknown. It is expressed at low levels in human placenta and shows restricted expression in lung endothelia, skin, liver, and kidney. The gene is also found to be selectively overexpressed in acute 203074_at ANXA8 244 602396 annexin A8 myelocytic leukemia. -

Aneuploidy: Using Genetic Instability to Preserve a Haploid Genome?
Health Science Campus FINAL APPROVAL OF DISSERTATION Doctor of Philosophy in Biomedical Science (Cancer Biology) Aneuploidy: Using genetic instability to preserve a haploid genome? Submitted by: Ramona Ramdath In partial fulfillment of the requirements for the degree of Doctor of Philosophy in Biomedical Science Examination Committee Signature/Date Major Advisor: David Allison, M.D., Ph.D. Academic James Trempe, Ph.D. Advisory Committee: David Giovanucci, Ph.D. Randall Ruch, Ph.D. Ronald Mellgren, Ph.D. Senior Associate Dean College of Graduate Studies Michael S. Bisesi, Ph.D. Date of Defense: April 10, 2009 Aneuploidy: Using genetic instability to preserve a haploid genome? Ramona Ramdath University of Toledo, Health Science Campus 2009 Dedication I dedicate this dissertation to my grandfather who died of lung cancer two years ago, but who always instilled in us the value and importance of education. And to my mom and sister, both of whom have been pillars of support and stimulating conversations. To my sister, Rehanna, especially- I hope this inspires you to achieve all that you want to in life, academically and otherwise. ii Acknowledgements As we go through these academic journeys, there are so many along the way that make an impact not only on our work, but on our lives as well, and I would like to say a heartfelt thank you to all of those people: My Committee members- Dr. James Trempe, Dr. David Giovanucchi, Dr. Ronald Mellgren and Dr. Randall Ruch for their guidance, suggestions, support and confidence in me. My major advisor- Dr. David Allison, for his constructive criticism and positive reinforcement. -

Pan-Genome-Wide Analysis of Pantoea Ananatis Identified Genes Linked to Pathogenicity In
bioRxiv preprint doi: https://doi.org/10.1101/2020.07.24.219337; this version posted July 24, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Pan-genome-wide analysis of Pantoea ananatis identified genes linked to pathogenicity in onion Gaurav Agarwal1*, Divya Choudhary1, Shaun P. Stice2, Brendon K. Myers1, Ronald D. Gitaitis1, Stephanus N. Venter3, Brian H. Kvitko2, Bhabesh Dutta1* 1Department of Plant Pathology, Coastal Plain Experimental Station, University of Georgia, Tifton, Georgia, USA 2Department of Plant Pathology, University of Georgia, Athens, Georgia, USA 3Department of Biochemistry, Genetics and Microbiology, Forestry and Agricultural Biotechnology Institute, University of Pretoria, Pretoria, South Africa *Corresponding authors: [email protected] (BD); [email protected] (GA) 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.07.24.219337; this version posted July 24, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Abstract Pantoea ananatis is a member of a Pantoea spp. complex that causes center rot of onion, which significantly affects onion yield and quality. This pathogen does not have typical virulence factors like type II or type III secretion systems but appears to require a biosynthetic gene-cluster, HiVir/PASVIL (located chromosomally), for a phosphonate secondary metabolite, and the onion- virulence regions, OVR (localized on a megaplasmid), for onion pathogenicity and virulence, respectively. -

Introgression in Betula Species of Different Ploidy Levels and the Analysis of the Betula Nana Genome
Introgression in Betula Species of Different Ploidy Levels and the Analysis of the Betula nana Genome JASMIN ZOHREN School of Biological and Chemical Sciences Queen Mary University of London Mile End Road London E1 4NS Supervisors: Dr Richard J. A. Buggs Prof Richard A. Nichols November 2016 Submitted in partial fulfilment of the requirements of the Degree of Doctor of Philosophy 1 Statement of Originality I, Jasmin Zohren, confirm that the research included within this thesis is my own work or that where it has been carried out in collaboration with, or supported by others, that this is duly acknowledged below and my contribution indicated. Previously published material is also acknowledged below. I attest that I have exercised reasonable care to ensure that the work is original, and does not to the best of my knowledge break any UK law, infringe any third party’s copyright or other Intellectual Property Right, or contain any confidential material. I accept that the College has the right to use plagiarism detection software to check the electronic version of the thesis. I confirm that this thesis has not been previously submitted for the award of a degree by this or any other university. The copyright of this thesis rests with the author and no quotation from it or information derived from it may be published without the prior written consent of the author. Signature: Date: Details of collaboration and publications Chapter 2 is published in Zohren et al. (2016): Zohren J, Wang N, Kardailsky I, Borrell JS, Joecker A, Nichols RA, Buggs RJA (2016). -

Workflows for Rapid Functional Annotation of Diverse
insects Article Workflows for Rapid Functional Annotation of Diverse Arthropod Genomes Surya Saha 1,2 , Amanda M. Cooksey 2,3, Anna K. Childers 4 , Monica F. Poelchau 5 and Fiona M. McCarthy 2,* 1 Boyce Thompson Institute, 533 Tower Rd., Ithaca, NY 14853, USA; [email protected] 2 School of Animal and Comparative Biomedical Sciences, University of Arizona, 1117 E. Lowell St., Tucson, AZ 85721, USA; [email protected] 3 CyVerse, BioScience Research Laboratories, University of Arizona, 1230 N. Cherry Ave., Tucson, AZ 85721, USA 4 Bee Research Laboratory, Beltsville Agricultural Research Center, Agricultural Research Service, USDA, 10300 Baltimore Ave., Beltsville, MD 20705, USA; [email protected] 5 National Agricultural Library, Agricultural Research Service, USDA, 10301 Baltimore Ave., Beltsville, MD 20705, USA; [email protected] * Correspondence: fi[email protected] Simple Summary: Genomic technologies are accumulating information about genes faster than ever before, and sequencing initiatives, such as the Earth BioGenome Project, i5k, and Ag100Pest Initiative, are expected to increase this rate of acquisition. However, if genomic sequencing is to be used for the improvement of human health, agriculture, and our understanding of biological systems, it is necessary to identify genes and understand how they contribute to biological outcomes. While there are several well-established workflows for assembling genomic sequences and identifying genes, understanding gene function is essential to create actionable knowledge. Moreover, this functional annotation process must be easily accessible and provide information at a genomic scale to keep up Citation: Saha, S.; Cooksey, A.M.; with new sequence data. We report a well-defined workflow for rapid functional annotation of whole Childers, A.K.; Poelchau, M.F.; proteomes to produce Gene Ontology and pathways information. -

Microsatellites As EWS/FLI Response Elements in Ewing's Sarcoma
Microsatellites as EWS/FLI response elements in Ewing’s sarcoma Kunal Gangwal*†, Savita Sankar*†, Peter C. Hollenhorst*†, Michelle Kinsey*†, Stephen C. Haroldsen†, Atul A. Shah†, Kenneth M. Boucher*†, W. Scott Watkins‡, Lynn B. Jorde‡, Barbara J. Graves*†, and Stephen L. Lessnick*†§¶ʈ *Department of Oncological Sciences, †Huntsman Cancer Institute, ‡Department of Human Genetics, §Huntsman Cancer Institute Center for Children, and ¶Division of Pediatric Hematology/Oncology, University of Utah, Salt Lake City, UT 84112 Edited by Robert N. Eisenman, Fred Hutchinson Cancer Research Center, Seattle, WA, and approved May 3, 2008 (received for review February 4, 2008) The ETS gene family is frequently involved in chromosome trans- in close proximity to low affinity binding sites for other tran- locations that cause human cancer, including prostate cancer, scription factors that allow for cooperative DNA binding. leukemia, and sarcoma. However, the mechanisms by which on- Ewing’s sarcoma was the first tumor in which ETS family cogenic ETS proteins, which are DNA-binding transcription factors, members were shown to be involved in chromosomal transloca- target genes necessary for tumorigenesis is not well understood. tions and serves as a paradigm for ETS-driven cancers (10). Ewing’s sarcoma serves as a paradigm for the entire class of Ewing’s sarcoma is a highly malignant solid tumor of children ETS-associated tumors because nearly all cases harbor recurrent and young adults that usually harbors a recurrent chromosomal chromosomal translocations involving ETS genes. The most com- translocation, t(11;22)(q24;q12), that encodes the EWS/FLI mon translocation in Ewing’s sarcoma encodes the EWS/FLI onco- fusion oncoprotein (10).