Transcription Factors and Cis-Acting Elements in T Helper Cell
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Theory of an Immune System Retrovirus
Proc. Nati. Acad. Sci. USA Vol. 83, pp. 9159-9163, December 1986 Medical Sciences Theory of an immune system retrovirus (human immunodeficiency virus/acquired immune deficiency syndrome) LEON N COOPER Physics Department and Center for Neural Science, Brown University, Providence, RI 02912 Contributed by Leon N Cooper, July 23, 1986 ABSTRACT Human immunodeficiency virus (HIV; for- initiates clonal expansion, sustained by interleukin 2 and y merly known as human T-cell lymphotropic virus type interferon. Ill/lymphadenopathy-associated virus, HTLV-Ill/LAV), the I first give a brief sketch of these events in a linked- retrovirus that infects T4-positive (helper) T cells of the interaction model in which it is assumed that antigen-specific immune system, has been implicated as the agent responsible T cells must interact with the B-cell-processed virus to for the acquired immune deficiency syndrome. In this paper, initiate clonal expansion (2). I then assume that virus-specific I contrast the growth of a "normal" virus with what I call an antibody is the major component ofimmune system response immune system retrovirus: a retrovirus that attacks the T4- that limits virus spread. As will be seen, the details of these positive T cells of the immune system. I show that remarkable assumptions do not affect the qualitative features of my interactions with other infections as well as strong virus conclusions. concentration dependence are general properties of immune Linked-Interaction Model for Clonal Expansion of Lympho- system retroviruses. Some of the consequences of these ideas cytes. Let X be the concentration of normal infecting virus are compared with observations. -

Cells Through Regulation of Follicular Helper T B7RP-1 Blockade
The Journal of Immunology B7RP-1 Blockade Ameliorates Autoimmunity through Regulation of Follicular Helper T Cells Yi-Ling Hu,* Daniela P. Metz,* James Chung,† Gerald Siu,1 and Ming Zhang2* Autoimmune diseases are marked by the presence of class-switched, high-affinity autoantibodies with pathogenic potential. Co- stimulation plays an important role in the activation of T cells and the development of T cell-dependent B cell responses. ICOS plays an indispensable role in the development of follicular helper T cells (TFH cells), which provide cognate help to germinal center (GC) B cells. We show that the levels of TFH cells and GC B cells in two different models of autoimmunity, the New Zealand Black/New Zealand White (NZB/NZW) F1 mouse model of systemic lupus erythematosus and the collagen-induced arthritis model of rheumatoid arthritis, are dependent on the maintenance of the ICOS/B7RP-1 pathway. Treatment with an anti-B7RP-1 Ab ameliorates disease manifestations and leads to a decrease in TFH cells and GC B cells as well as an overall decrease in the frequency of ICOS؉ T cells. Coculture experiments of Ag-primed B cells with CXCR5؉ or CXCR5؊ T cells show that blocking B7RP-1 does not directly impact the production of IgG by B cells. These findings further support the role of ICOS in autoimmunity and suggest that the expansion of the TFH cell pool is an important mechanism by which ICOS regulates Ab production. The Journal of Immunology, 2009, 182: 1421–1428. ffective activation of T cells via their Ag receptor requires ing anti-B7RP-1 reagents effectively inhibited the initiation and additional stimuli provided by cell-surface costimulatory maturation of an Ab-mediated Ag response (15). -

Influence of Vitamin C on Lymphocytes: an Overview
antioxidants Review Influence of Vitamin C on Lymphocytes: An Overview Gwendolyn N. Y. van Gorkom 1,*, Roel G. J. Klein Wolterink 1, Catharina H. M. J. Van Elssen 1 ID , Lotte Wieten 2, Wilfred T. V. Germeraad 1 and Gerard M. J. Bos 1 1 Division of Hematology, Department of Internal Medicine, GROW-School for Oncology and Developmental Biology, Maastricht University Medical Center, 6202AZ Maastricht, The Netherlands; [email protected] (R.G.J.K.W.); [email protected] (C.H.M.J.V.E.); [email protected] (W.T.V.G.); [email protected] (G.M.J.B.) 2 Department of Transplantation Immunology, Maastricht University Medical Center, 6202 AZ Maastricht, The Netherlands; [email protected] * Correspondence: [email protected]; Tel.: +31-043-387-6543 Received: 8 February 2018; Accepted: 8 March 2018; Published: 10 March 2018 Abstract: Vitamin C or ascorbic acid (AA) is implicated in many biological processes and has been proposed as a supplement for various conditions, including cancer. In this review, we discuss the effects of AA on the development and function of lymphocytes. This is important in the light of cancer treatment, as the immune system needs to regenerate following chemotherapy or stem cell transplantation, while cancer patients are often AA-deficient. We focus on lymphocytes, as these white blood cells are the slowest to restore, rendering patients susceptible to often lethal infections. T lymphocytes mediate cellular immunity and have been most extensively studied in the context of AA biology. In vitro studies demonstrate that T cell development requires AA, while AA also enhances T cell proliferation and may influence T cell function. -
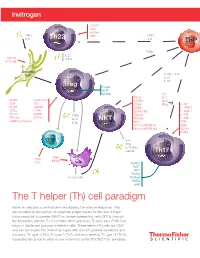
Cell Paradigm
CCR10 CD4 PDGFR TNFα Th22 TCR TNFα I L- 6 I L- 6 ThP TGFβ I L-2 2 CD11C TNFα DEC205 Dendritic cell TGFβ + IL-6 I L-21 I L-23 Treg Foxp3 AHR STAT5 I L-2 CD25 I L- 4 CD25 GITR/AITR CD49b IL-27 CD4 TCR CD69 IFNγ T I L-1R1 CD39 CXCR3 CD94 I L-12R B1 CD73 CCR4 CD160 (m) I L-13 R α1 CD101 CCR9 TGFβ CD244 I L-21R FR4 (m) CTLA-4 I L-10 NKT NKG2D I L-23 R GARP (activated) IL-35 NK1.1 TCR Vα24Jα18TCR (h) CD4 Vα14Jα18TCR (m) CD161 CCR6 Effector T I L- 4 IFNγ I L-17 ( i N K T subset) Th17 MHCI APC TCR MHCII CD3 RUNX1 BATF IRF4 RORγt I L-15 / I L-15 R RORα4 STAT3 AHR The T helper (Th) cell paradigm Naive Th cells play a central role in modulating the immune response. They are activated by recognition of a peptide antigen bound to the class II major histocompatibility complex (MHC) on antigen-presenting cells (APCs) through the interaction with the T cell receptor. After activation, Th precursor (ThP) cells begin to divide and give rise to effector cells. These effector Th cells are CD4⁺ and can be divided into three main types with distinct cytokine secretions and functions: Th type 1 (Th1), Th type 2 (Th2), and most recently Th type 17 (Th17), expanding the group to what is now referred to as the Th1/Th2/Th17 paradigm. The T helper (Th) cell paradigm DEC205 Dendritic CD11c cell CCR10 CD4 CD4 PDGFR TCR TNFα Th22 TCR TNFα Antigen presentation I L- 6 I L- 6 ThP Costimulatory signals TGFβ Th9 I L- 4 TGFβ I L-2 2 CD11C I L- 9 TNFα I L-2 I L-25 DEC205 I L- 4 IL-31 I L-15 ( h) IL-33 TCR CD4 Dendritic CD30 cell TGFβ + IL-6 I L-21 CCR8 I L-23 I L- 6 CXCR4 -

Harnessing the Plasticity of CD4+ T Cells to Treat Immune-Mediated Disease
REVIEWS Harnessing the plasticity of CD4+ T cells to treat immune-mediated disease Michel DuPage and Jeffrey A. Bluestone Abstract | CD4+ T cells differentiate and acquire distinct functions to combat specific pathogens but can also adapt their functions in response to changing circumstances. Although this phenotypic plasticity can be potentially deleterious, driving immune pathology, it also provides important benefits that have led to its evolutionary preservation. Here, we review CD4+ T cell plasticity by examining the molecular mechanisms that regulate it — from the extracellular cues that initiate and drive cells towards varying phenotypes, to the cytosolic signalling cascades that decipher these cues and transmit them into the cell and to the nucleus, where these signals imprint specific gene expression programmes. By understanding how this functional flexibility is achieved, we may open doors to new therapeutic approaches that harness this property of T cells. The hypothesis of T helper 1 (TH1) and TH2 cell subsets Thus, there has been a re‑emergence of T cell ‘plas‑ championed by Mosmann and Coffman in the 1980s, ticity’, as opposed to ‘lineage stability’, as the evolving and others since, provided a framework to understand paradigm12 (BOX 1). how CD4+ T cells direct diverse immune responses1,2. In this Review, we summarize a rapidly expanding By examining clonal populations of CD4+ T cells, they literature surrounding CD4+ T cell subsets and make found that different clones expressed selected pat‑ the case for broad plasticity -

T Helper Cell Differentiation: Understanding the Needs of Hierarchy
Immunity Previews T Helper Cell Differentiation: Understanding the Needs of Hierarchy Thomas Weichhart1,* and Marcus D. Sa¨ emann1,* 1Department of Internal Medicine III, Clinical Division of Nephrology and Dialysis, Medical University Vienna, Wa¨ hringer Gu¨ rtel 18-20, A-1090 Vienna, Austria *Correspondence: [email protected] (T.W.), [email protected] (M.D.S.) DOI 10.1016/j.immuni.2010.06.008 In this issue of Immunity, Lee et al. (2010) demonstrate that the mammalian Target of Rapamycin Complex 2 promotes the differentiation of T helper 1 (Th1) cells via the kinase Akt, whereas it independently fosters Th2 cell generation via another kinase, PKC-q. Regulation of the manifold and flexible mTOR is a critical regulator of memory Akt S473 was defective after stimulation requirements of an efficient immune CD8+ T cell generation as well as inflam- with CD3 and CD28 mAb, whereas response to eliminate dangerous micro- matory responses in myeloid antigen-pre- mTORC1-mediated phosphorylation of bes depends on a delicate balance of senting cells (Araki et al., 2009; Weichhart S6K1 was intact. Functionally, however, diverse T helper (Th) cell subsets (Zhu et al., 2008). mTOR is the core component it was found that Rictor-deficient CD4+ et al., 2010). Hence, naive CD4+ Th cells of mTORC1 (mTOR complex 1), which is T cells were unable to differentiate into differentiate into polarized effector Th composed by the adaptor protein Raptor, both Th1 or Th2 cells in vitro, demon- cell subsets depending on the priming whereas Rictor and Sin1 classify strating that mTORC2 is vital for Th1 and cytokine milieu. -

6229 T Cell Regulation of Hematopoiesis Alexander L Dent1
[Frontiers in Bioscience 13, 6229-6236, May 1, 2008] T cell regulation of hematopoiesis Alexander L Dent1, Mark H. Kaplan1,2 1Department of Microbiology and Immunology and Walther Oncology Center, Indiana University School of Medicine, Indianapolis, IN 46202, 2Department of Pediatrics, Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202 TABLE OF CONTENTS 1. Abstract 2. Introduction 3. T cells are required for myeloid cell maturation 4. Th1 cells regulate Hematopoietic Progenitor Cell homeostasis 5. The role of Th2 cells in modulating hematopoiesis 6. IL-17-secreting “Th17” cells regulate peripheral neutrophil numbers 7. Natural killer T cell regulate HSC and HPC activity 8. CD8 cells can alter hematopoietic progenitor cell activity. 9. Do Regulatory T cells influence hematopoiesis? 10. Perspective 11. References 1. ABSTRACT 2. INTRODUCTION It has long been known that thymus-derived lymphocytes (T cells) can produce cytokines that have T cells secrete several well-known cytokines, powerful effects on hematopoiesis. All major classes of T such as IL-3, IL-5, IL-6, GM-CSF and M-CSF that strongly cells-- CD4 T helper cells, CD4 regulatory T cells, CD8 T influence and promote hematopoietic cell development. cells, γδ T cells and NKT cells-- produce a number of These cytokines act on hematopoiesis through mechanisms cytokines and chemokines that can modulate that are both direct and indirect (1-3). Each of these hematopoiesis. More recent research has shown that cytokines can potentially regulate hematopoiesis at several specific T helper cell types, such as Th1, Th2 and Th17 stages, including the regulation of hematopoietic stem cells cells, with the development of each subset depending on (HSC), hematopoietic progenitor cells (HPC) and mature distinct STAT proteins, have the potential to modulate the cells in the periphery. -
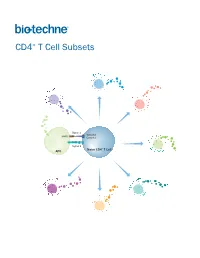
CD4+ T Cell Subsets
CD4+ T Cell Subsets Th2 Th1 Th9 Signal 1 TCR-CD3 MHCII Complex Th17 Signal 2 Naïve CD4+ T Cell APC Treg Th22 Tfh CD4+ T Cell Subsets CD4+ T cells play a central role in directing adaptive immune responses against invading microbial pathogens. Naïve CD4+ T cells differentiate into more specialized subsets following T cell activation, which requires recognition of the antigenic peptide/major histocompatibility complex (MHC) by the T cell receptor (Signal 1), along with antigen-independent co-stimulation (Signal 2). Differentiation into a specific CD4+ T cell subset depends on the antigen, the strength of the TCR signal, and cytokines present in the surrounding extracellular environment. The different CD4+ T helper cell subsets that have been identified include T helper 1 (Th1), Th2, Th9, Th17, and Th22 cells, along with follicular helper T (Tfh) cells, and regulatory T cells (Tregs). Each of these subsets expresses unique combinations of cell surface receptors, transcription factors, and secreted cytokines. While Tfh cells are primarily involved in promoting the survival and proliferation of germinal center B cells and supporting germinal center development, the Th1, Th2, Th9, Th17, and Th22 subsets have all been found to be involved in host defense against specific microbial pathogens. The defined array of cytokines secreted by these effector T cell subsets contributes to both elimination of the foreign pathogen and activation of other immune cell types. The activities of these cells are balanced in part by a unique subpopulation of CD4+ T cells known as regulatory T cells (Tregs). Tregs specialize in maintaining immune homeostasis and self-tolerance, dampening inflammation, and preventing the development of autoimmune disease. -

The Five Elements of Th1-Th2 System
The Five Elements of Th1-Th2 System Shengrong Zou* Information Engineering College, Yangzhou University , Yangzhou 225009 JiangSu ,PR China __________________________________________________________________________________________ Abstract The T helper (Th) phenotypes, Th1/Th2, are acquired upon interaction of a naive T helper cell and an antigen presenting cell (APC). Naive T helper cells may differentiate into either phenotype, and the actual outcome is determined by the density and avidity of the antigenic determinants presented by the APC, and the APCs inherent costimulatory properties. Until recently it was thought that differentiation is further affected by cytokines. In our work, We have specified aspects of T-cell cytokine networks using B method of software engineering. With this model, we are able to run verification with B-toolkit and allow us to compare the dynamic behavior of the model to actual experimental data from College of Animal Science and Veterinary Medicine. Here we present a Wu-Hsing model of Th1-Th2 system. Keywords: T cell ; Cytokine network ; Th1-Th2 system; Wu-Hsing __________________________________________________________________________________________ Contents 1. Introduction……………………………………………….……………………………………………..……………………...1 2. The Wu-Hsing model…….…………………..…………………………………….……………………..……………….…….1 3. The five elements of Th1-Th2 system……..………………………. …………….………….….…………………….….2 3.1 Five elements……………………………………………………………….…………………………………………….3 3.2 Promotion……………………………………………………………………………………………….………………..3 3.3 Inhibition…………………………………………………………………………………………………………………4 -

Hematopoiesis and T-Cell Specification As a Model Developmental System
View metadata, citation and similar papers at core.ac.uk brought to you by CORE HHS Public Access provided by Caltech Authors - Main Author manuscript Author ManuscriptAuthor Manuscript Author Immunol Manuscript Author Rev. Author manuscript; Manuscript Author available in PMC 2017 May 01. Published in final edited form as: Immunol Rev. 2016 May ; 271(1): 72–97. doi:10.1111/imr.12417. HEMATOPOIESIS AND T-CELL SPECIFICATION AS A MODEL DEVELOPMENTAL SYSTEM Ellen V. Rothenberg*, Hao Yuan Kueh, Mary A. Yui, and Jingli A. Zhang† Division of Biology & Biological Engineering, California Institute of Technology, Pasadena, CA 91125 USA SUMMARY The pathway to generate T cells from hematopoietic stem cells guides progenitors through a succession of fate choices while balancing differentiation progression against proliferation, stage to stage. Many elements of the regulatory system that controls this process are known, but the requirement for multiple, functionally distinct transcription factors needs clarification in terms of gene network architecture. Here we compare the features of the T-cell specification system with the rule sets underlying two other influential types of gene network models: first, the combinatorial, hierarchical regulatory systems that generate the orderly, synchronized increases in complexity in most invertebrate embryos; second, the dueling “master regulator” systems that are commonly used to explain bistability in microbial systems and in many fate choices in terminal differentiation. The T-cell specification process shares certain features with each of these prevalent models but differs from both of them in central respects. The T-cell system is highly combinatorial but also highly dose-sensitive in its use of crucial regulatory factors. -

Based Vaccination and Nonviral Cytokine Gene Transfer
Gene Therapy (2000) 7, 481–492 2000 Macmillan Publishers Ltd All rights reserved 0969-7128/00 $15.00 www.nature.com/gt NONVIRAL TRANSFER TECHNOLOGY RESEARCH ARTICLE Regulation of T-helper-1 versus T-helper-2 activity and enhancement of tumor immunity by combined DNA- based vaccination and nonviral cytokine gene transfer K Song, Y Chang, and GJ Prud’homme Department of Pathology, McGill University, 3775 University Street, Room 13, Montreal, Quebec, Canada, H3A 2B4 Intramuscular (i.m.) injections of a plasmid encoding human type 2 (Th2) response. Antitumor immunity was enhanced carcinoembryonic antigen (CEA) elicited both humoral and when the CEA and IL-12 plasmids were coinjected at the cellular immune responses in mice, but only partial inhibition same muscle site, but not at separate sites despite of the growth of transplanted syngeneic CEA-positive P815 increased serum IL-12 levels. Though the tumor cells tumor cells (CEA/P815). Coinjection of the CEA vector with a expressed neomycin phosphotransferase, mice immunized vector encoding either interferon-␥ (IFN␥) or IL-12 promoted with vectors encoding that protein (without CEA) were not IgG2a isotype anti-CEA antibody production, anti-CEA/P815 protected against tumor growth, and produced no CTLs CTL activity and greater resistance to CEA/P815 tumor chal- except for low levels when coinjected with an IL-12 vector. lenge. As well, CEA/P815-stimulated IFN␥ secretion in vitro Thus, we show that immunity elicited by DNA vaccination was increased, but IL-4 diminished, consistent with a T- against CEA can be biased to a protective type (high Th1 helper type 1 (Th1) response. -

The Darker Side of Follicular Helper T Cells: from Autoimmunity to Immunodeficiency
Cellular & Molecular Immunology (2012) 9, 380–385 ß 2012 CSI and USTC. All rights reserved 1672-7681/12 $32.00 www.nature.com/cmi REVIEW The darker side of follicular helper T cells: from autoimmunity to immunodeficiency Sudhanshu Shekhar and Xi Yang 1 Follicular helper T (TFH) cells represent a distinct subset of CD4 helper T (TH) cells specialized in providing help to B cells. They are characterized by their unique transcriptional profile (Bcl6), surface marker expression (CXCR5, PD-1, ICOS and CD40L) and cytokine production pattern (IL-21 and IL-6). TFH cells provide help to B cells both to form germinal centers (GCs) and to differentiate into memory B cells and plasma cells for generation of humoral responses. However, there is emerging evidence that implicates TFH cells in the development of various human pathologies, such as autoimmune diseases, immunodeficiency and lymphoma. This review focuses on the current progress in this area including mouse and human studies. A clearer understanding of the mechanisms of TFH cell-mediated immunity and pathology may be exploited for rational development of therapeutic strategies. Cellular & Molecular Immunology (2012) 9, 380–385; doi:10.1038/cmi.2012.26; published online 13 August 2012 Keywords: autoimmunity; B cells; immunodeficiency; lymphoma; TFH cells 13,14 INTRODUCTION TFH cells to B-cell follicles for their interaction with B cells. Upon Antibody responses are a key component of the adaptive immune TFH-cell activation, PD-1 induces an inhibitory signal to TFH cells, system. These responses are mostly dependent on help from anti- whereas ICOS functions as a costimulatory molecule, thus determin- 1 15,16 gen-specific CD4 helper T (TH) cells.