Hematopoiesis and T-Cell Specification As a Model Developmental System
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

ILC2 Activation by Protozoan Commensal Microbes
International Journal of Molecular Sciences Review ILC2 Activation by Protozoan Commensal Microbes Kyle Burrows 1 , Louis Ngai 1 , Flora Wong 1,2, David Won 1 and Arthur Mortha 1,* 1 University of Toronto, Department of Immunology, Toronto, ON M5S 1A8, Canada; [email protected] (K.B.) [email protected] (L.N.); fl[email protected] (F.W.); [email protected] (D.W.) 2 Ranomics, Inc. Toronto, ON M5G 1X5, Canada * Correspondence: [email protected] Received: 3 September 2019; Accepted: 27 September 2019; Published: 30 September 2019 Abstract: Group 2 innate lymphoid cells (ILC2s) are a member of the ILC family and are involved in protective and pathogenic type 2 responses. Recent research has highlighted their involvement in modulating tissue and immune homeostasis during health and disease and has uncovered critical signaling circuits. While interactions of ILC2s with the bacterial microbiome are rather sparse, other microbial members of our microbiome, including helminths and protozoans, reveal new and exciting mechanisms of tissue regulation by ILC2s. Here we summarize the current field on ILC2 activation by the tissue and immune environment and highlight particularly new intriguing pathways of ILC2 regulation by protozoan commensals in the intestinal tract. Keywords: ILC2; protozoa; Trichomonas; Tritrichomonas musculis; mucosal immunity; taste receptors; succinate; intestinal immunity; type 2 immunity; commensals 1. The ILC Lineage 1.1. The Family of Innate Lymphoid Cells Research over the last decade has redirected focus away from classical immune cell interactions within lymphoid tissues towards immunity within non-lymphoid tissues. Within these tissues, immune interactions involve local adaptation and rapid responses by tissue-resident immune cells. -

Theory of an Immune System Retrovirus
Proc. Nati. Acad. Sci. USA Vol. 83, pp. 9159-9163, December 1986 Medical Sciences Theory of an immune system retrovirus (human immunodeficiency virus/acquired immune deficiency syndrome) LEON N COOPER Physics Department and Center for Neural Science, Brown University, Providence, RI 02912 Contributed by Leon N Cooper, July 23, 1986 ABSTRACT Human immunodeficiency virus (HIV; for- initiates clonal expansion, sustained by interleukin 2 and y merly known as human T-cell lymphotropic virus type interferon. Ill/lymphadenopathy-associated virus, HTLV-Ill/LAV), the I first give a brief sketch of these events in a linked- retrovirus that infects T4-positive (helper) T cells of the interaction model in which it is assumed that antigen-specific immune system, has been implicated as the agent responsible T cells must interact with the B-cell-processed virus to for the acquired immune deficiency syndrome. In this paper, initiate clonal expansion (2). I then assume that virus-specific I contrast the growth of a "normal" virus with what I call an antibody is the major component ofimmune system response immune system retrovirus: a retrovirus that attacks the T4- that limits virus spread. As will be seen, the details of these positive T cells of the immune system. I show that remarkable assumptions do not affect the qualitative features of my interactions with other infections as well as strong virus conclusions. concentration dependence are general properties of immune Linked-Interaction Model for Clonal Expansion of Lympho- system retroviruses. Some of the consequences of these ideas cytes. Let X be the concentration of normal infecting virus are compared with observations. -

Innate Lymphocytes—Lineage, Localization and Timing of Differentiation
Cellular & Molecular Immunology www.nature.com/cmi REVIEW ARTICLE Innate lymphocytes—lineage, localization and timing of differentiation Emily R. Kansler1,2 and Ming O. Li1 Innate lymphocytes are a diverse population of cells that carry out specialized functions in steady-state homeostasis and during immune challenge. While circulating cytotoxic natural killer (NK) cells have been studied for decades, tissue-resident innate lymphoid cells (ILCs) have only been characterized and studied over the past few years. As ILCs have been largely viewed in the context of helper T-cell biology, models of ILC lineage and function have been founded within this perspective. Notably, tissue- resident innate lymphocytes with cytotoxic potential have been described in an array of tissues, yet whether they are derived from the NK or ILC lineage is only beginning to be elucidated. In this review, we aim to shed light on the identities of innate lymphocytes through the lenses of cell lineage, localization, and timing of differentiation. Cellular & Molecular Immunology (2019) 16:627–633; https://doi.org/10.1038/s41423-019-0211-7 INTRODUCTION memory T cells have been described.3 Central memory T (Tcm) Lymphocytes are a fundamental component of the host immune cells are derived from CX3CR1− effector T cells and primarily response to challenge. Different classes of the lymphocyte circulate between the blood and secondary lymphoid organs. response are best defined by the type of effector programs Resident memory T (Trm) cells, although also derived from carried out by CD8+ or CD4+ T cells. CD8+ T cells are cytotoxic CX3CR1− effector T cells, enter peripheral tissues where they are lymphocytes that mediate a type 1 immune response to maintained locally by self-renewal and do not recirculate. -

Cells Through Regulation of Follicular Helper T B7RP-1 Blockade
The Journal of Immunology B7RP-1 Blockade Ameliorates Autoimmunity through Regulation of Follicular Helper T Cells Yi-Ling Hu,* Daniela P. Metz,* James Chung,† Gerald Siu,1 and Ming Zhang2* Autoimmune diseases are marked by the presence of class-switched, high-affinity autoantibodies with pathogenic potential. Co- stimulation plays an important role in the activation of T cells and the development of T cell-dependent B cell responses. ICOS plays an indispensable role in the development of follicular helper T cells (TFH cells), which provide cognate help to germinal center (GC) B cells. We show that the levels of TFH cells and GC B cells in two different models of autoimmunity, the New Zealand Black/New Zealand White (NZB/NZW) F1 mouse model of systemic lupus erythematosus and the collagen-induced arthritis model of rheumatoid arthritis, are dependent on the maintenance of the ICOS/B7RP-1 pathway. Treatment with an anti-B7RP-1 Ab ameliorates disease manifestations and leads to a decrease in TFH cells and GC B cells as well as an overall decrease in the frequency of ICOS؉ T cells. Coculture experiments of Ag-primed B cells with CXCR5؉ or CXCR5؊ T cells show that blocking B7RP-1 does not directly impact the production of IgG by B cells. These findings further support the role of ICOS in autoimmunity and suggest that the expansion of the TFH cell pool is an important mechanism by which ICOS regulates Ab production. The Journal of Immunology, 2009, 182: 1421–1428. ffective activation of T cells via their Ag receptor requires ing anti-B7RP-1 reagents effectively inhibited the initiation and additional stimuli provided by cell-surface costimulatory maturation of an Ab-mediated Ag response (15). -

Influence of Vitamin C on Lymphocytes: an Overview
antioxidants Review Influence of Vitamin C on Lymphocytes: An Overview Gwendolyn N. Y. van Gorkom 1,*, Roel G. J. Klein Wolterink 1, Catharina H. M. J. Van Elssen 1 ID , Lotte Wieten 2, Wilfred T. V. Germeraad 1 and Gerard M. J. Bos 1 1 Division of Hematology, Department of Internal Medicine, GROW-School for Oncology and Developmental Biology, Maastricht University Medical Center, 6202AZ Maastricht, The Netherlands; [email protected] (R.G.J.K.W.); [email protected] (C.H.M.J.V.E.); [email protected] (W.T.V.G.); [email protected] (G.M.J.B.) 2 Department of Transplantation Immunology, Maastricht University Medical Center, 6202 AZ Maastricht, The Netherlands; [email protected] * Correspondence: [email protected]; Tel.: +31-043-387-6543 Received: 8 February 2018; Accepted: 8 March 2018; Published: 10 March 2018 Abstract: Vitamin C or ascorbic acid (AA) is implicated in many biological processes and has been proposed as a supplement for various conditions, including cancer. In this review, we discuss the effects of AA on the development and function of lymphocytes. This is important in the light of cancer treatment, as the immune system needs to regenerate following chemotherapy or stem cell transplantation, while cancer patients are often AA-deficient. We focus on lymphocytes, as these white blood cells are the slowest to restore, rendering patients susceptible to often lethal infections. T lymphocytes mediate cellular immunity and have been most extensively studied in the context of AA biology. In vitro studies demonstrate that T cell development requires AA, while AA also enhances T cell proliferation and may influence T cell function. -
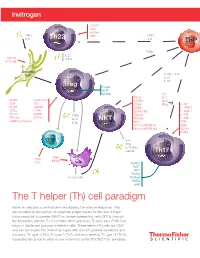
Cell Paradigm
CCR10 CD4 PDGFR TNFα Th22 TCR TNFα I L- 6 I L- 6 ThP TGFβ I L-2 2 CD11C TNFα DEC205 Dendritic cell TGFβ + IL-6 I L-21 I L-23 Treg Foxp3 AHR STAT5 I L-2 CD25 I L- 4 CD25 GITR/AITR CD49b IL-27 CD4 TCR CD69 IFNγ T I L-1R1 CD39 CXCR3 CD94 I L-12R B1 CD73 CCR4 CD160 (m) I L-13 R α1 CD101 CCR9 TGFβ CD244 I L-21R FR4 (m) CTLA-4 I L-10 NKT NKG2D I L-23 R GARP (activated) IL-35 NK1.1 TCR Vα24Jα18TCR (h) CD4 Vα14Jα18TCR (m) CD161 CCR6 Effector T I L- 4 IFNγ I L-17 ( i N K T subset) Th17 MHCI APC TCR MHCII CD3 RUNX1 BATF IRF4 RORγt I L-15 / I L-15 R RORα4 STAT3 AHR The T helper (Th) cell paradigm Naive Th cells play a central role in modulating the immune response. They are activated by recognition of a peptide antigen bound to the class II major histocompatibility complex (MHC) on antigen-presenting cells (APCs) through the interaction with the T cell receptor. After activation, Th precursor (ThP) cells begin to divide and give rise to effector cells. These effector Th cells are CD4⁺ and can be divided into three main types with distinct cytokine secretions and functions: Th type 1 (Th1), Th type 2 (Th2), and most recently Th type 17 (Th17), expanding the group to what is now referred to as the Th1/Th2/Th17 paradigm. The T helper (Th) cell paradigm DEC205 Dendritic CD11c cell CCR10 CD4 CD4 PDGFR TCR TNFα Th22 TCR TNFα Antigen presentation I L- 6 I L- 6 ThP Costimulatory signals TGFβ Th9 I L- 4 TGFβ I L-2 2 CD11C I L- 9 TNFα I L-2 I L-25 DEC205 I L- 4 IL-31 I L-15 ( h) IL-33 TCR CD4 Dendritic CD30 cell TGFβ + IL-6 I L-21 CCR8 I L-23 I L- 6 CXCR4 -

Harnessing the Plasticity of CD4+ T Cells to Treat Immune-Mediated Disease
REVIEWS Harnessing the plasticity of CD4+ T cells to treat immune-mediated disease Michel DuPage and Jeffrey A. Bluestone Abstract | CD4+ T cells differentiate and acquire distinct functions to combat specific pathogens but can also adapt their functions in response to changing circumstances. Although this phenotypic plasticity can be potentially deleterious, driving immune pathology, it also provides important benefits that have led to its evolutionary preservation. Here, we review CD4+ T cell plasticity by examining the molecular mechanisms that regulate it — from the extracellular cues that initiate and drive cells towards varying phenotypes, to the cytosolic signalling cascades that decipher these cues and transmit them into the cell and to the nucleus, where these signals imprint specific gene expression programmes. By understanding how this functional flexibility is achieved, we may open doors to new therapeutic approaches that harness this property of T cells. The hypothesis of T helper 1 (TH1) and TH2 cell subsets Thus, there has been a re‑emergence of T cell ‘plas‑ championed by Mosmann and Coffman in the 1980s, ticity’, as opposed to ‘lineage stability’, as the evolving and others since, provided a framework to understand paradigm12 (BOX 1). how CD4+ T cells direct diverse immune responses1,2. In this Review, we summarize a rapidly expanding By examining clonal populations of CD4+ T cells, they literature surrounding CD4+ T cell subsets and make found that different clones expressed selected pat‑ the case for broad plasticity -

Blocking IL-25 Signalling Protects Against Gut Inflammation in a Type-2
J Gastroenterol (2012) 47:1198–1211 DOI 10.1007/s00535-012-0591-2 ORIGINAL ARTICLE—ALIMENTARY TRACT Blocking IL-25 signalling protects against gut inflammation in a type-2 model of colitis by suppressing nuocyte and NKT derived IL-13 Ana Camelo • Jillian L. Barlow • Lesley F. Drynan • Daniel R. Neill • Sarah J. Ballantyne • See Heng Wong • Richard Pannell • Wei Gao • Keely Wrigley • Justin Sprenkle • Andrew N. J. McKenzie Received: 18 November 2011 / Accepted: 22 March 2012 / Published online: 27 April 2012 Ó The Author(s) 2012. This article is published with open access at Springerlink.com Abstract infiltrates were assessed for mucosal inflammation and Background Interleukin-25 (IL-25) is a potent activator cultured in vitro to determine cytokine production. of type-2 immune responses. Mucosal inflammation in Results We found that in oxazolone colitis IL-25 pro- ulcerative colitis is driven by type-2 cytokines. We have duction derives from intestinal epithelial cells and that previously shown that a neutralizing anti-IL-25 antibody IL-17BR? IL-13-producing natural killer T (NKT) cells abrogated airways hyperreactivity in an experimental and nuocytes drive the intestinal inflammation. Blocking model of lung allergy. Therefore, we asked whether IL-25 signalling considerably improved the clinical aspects blocking IL-25 via neutralizing antibodies against the of the disease, including weight loss and colon ulceration, ligand or its receptor IL-17BR could protect against and resulted in fewer nuocytes and NKT cells infiltrating inflammation in an oxazolone-induced mouse model of the mucosa. The improved pathology correlated with a colitis. decrease in IL-13 production by lamina propria cells, a Methods Neutralizing antibodies to IL-25 or IL-17BR decrease in the production of other type-2 cytokines by were administered to mice with oxazolone-induced colitis, MLN cells, and a decrease in blood eosinophilia and IgE. -

T Helper Cell Differentiation: Understanding the Needs of Hierarchy
Immunity Previews T Helper Cell Differentiation: Understanding the Needs of Hierarchy Thomas Weichhart1,* and Marcus D. Sa¨ emann1,* 1Department of Internal Medicine III, Clinical Division of Nephrology and Dialysis, Medical University Vienna, Wa¨ hringer Gu¨ rtel 18-20, A-1090 Vienna, Austria *Correspondence: [email protected] (T.W.), [email protected] (M.D.S.) DOI 10.1016/j.immuni.2010.06.008 In this issue of Immunity, Lee et al. (2010) demonstrate that the mammalian Target of Rapamycin Complex 2 promotes the differentiation of T helper 1 (Th1) cells via the kinase Akt, whereas it independently fosters Th2 cell generation via another kinase, PKC-q. Regulation of the manifold and flexible mTOR is a critical regulator of memory Akt S473 was defective after stimulation requirements of an efficient immune CD8+ T cell generation as well as inflam- with CD3 and CD28 mAb, whereas response to eliminate dangerous micro- matory responses in myeloid antigen-pre- mTORC1-mediated phosphorylation of bes depends on a delicate balance of senting cells (Araki et al., 2009; Weichhart S6K1 was intact. Functionally, however, diverse T helper (Th) cell subsets (Zhu et al., 2008). mTOR is the core component it was found that Rictor-deficient CD4+ et al., 2010). Hence, naive CD4+ Th cells of mTORC1 (mTOR complex 1), which is T cells were unable to differentiate into differentiate into polarized effector Th composed by the adaptor protein Raptor, both Th1 or Th2 cells in vitro, demon- cell subsets depending on the priming whereas Rictor and Sin1 classify strating that mTORC2 is vital for Th1 and cytokine milieu. -

The Role of Inhibitor of Binding Or Differentiation 2 in the Development
Immunobiology 224 (2019) 142–146 Contents lists available at ScienceDirect Immunobiology journal homepage: www.elsevier.com/locate/imbio Review The role of inhibitor of binding or differentiation 2 in the development and differentiation of immune cells T ⁎ Haojie Wua,b, Qixiang Shaoa,b, a Reproductive Sciences Institute of Jiangsu University, Zhenjiang 212001, Jiangsu, P.R. China b Department of Immunology, Key Laboratory of Medical Science and Laboratory Medicine of Jiangsu Province, School of Medicine, Jiangsu University, Zhenjiang 212013, Jiangsu, P.R. China ARTICLE INFO ABSTRACT Keywords: Inhibitor of binding or differentiation 2 (Id2), a member of helix-loop-helix (HLH) transcriptional factors, is Inhibitor of binding or differentiation 2 recently reported as an important regulator of the development or differentiation of immune cells. It has been T cell demonstrated that Id2 plays a critical role in the early lymphopoiesis. However, it has been discovered recently B cell that Id2 displays new functions in different immune cells. In the adaptive immune cells, Id2 is required for Dendritic cell determining T-cell subsets and B cells. In addition, Id2 is also involved in the development of innate immune Innate lymphoid cell cells, including dendritic cells (DCs), natural killer (NK) cells, and other innate lymphoid cells (ILCs). Here, we review the current reports about the role of Id2 in the development or differentiation of main immune cells. 1. Introduction dimerization when recognizing their targets (Ellenberger et al., 1994). Whereas, Id proteins lack this basic region (Benezra et al., 1990) The inhibitor of DNA binding or differentiation (Id) is a member of (Fig. 1). -

Programming for T-Lymphocyte Fates: Modularity and Mechanisms
Downloaded from genesdev.cshlp.org on October 4, 2021 - Published by Cold Spring Harbor Laboratory Press REVIEW Programming for T-lymphocyte fates: modularity and mechanisms Ellen V. Rothenberg Division of Biology and Biological Engineering, California Institute of Technology, Pasadena, California 91125, USA T-cell development in mammals is a model for lineage heritable—and often as irreversible—as true lineage choice and differentiation from multipotent stem cells. choices. Furthermore, their developmental path from Although T-cell fate choice is promoted by signaling in stem and progenitor cells is protracted compared with the thymus through one dominant pathway, the Notch other hematopoietic lineages and requires a specialized pathway, it entails a complex set of gene regulatory net- microenvironment; namely, the thymus. work and chromatin state changes even before the cells Major features of T-cell development appear to be con- begin to express their signature feature, the clonal-specific served among all jawed vertebrates (Litman et al. 2010), al- T-cell receptors (TCRs) for antigen. This review distin- beit with some variations, and it has become clear in the guishes three developmental modules for T-cell develop- last decade that a version of T-cell development also oc- ment, which correspond to cell type specification, TCR curs in the agnathan lamprey (Bajoghli et al. 2011). The expression and selection, and the assignment of cells to thymus itself is more phylogenetically conserved than different effector types. The first is based on transcription- the tissues that provide microenvironments for blood de- al regulatory network events, the second is dominated by velopment generally (Zapata and Amemiya 2000; Boehm somatic gene rearrangement and mutation and cell selec- and Bleul 2007). -

"Cd1d-Restricted Natural Killer T Cells" In
CD1d-Restricted Natural Advanced article Article Contents Killer T Cells • Introduction • CD1d Structure Dictates Lipid Ligand Timothy M Hill, Department of Pathology, Microbiology and Immunology, Van- Presentation derbilt University School of Medicine, Nashville, Tennessee, USA and Department of Chem- • NKT-Cell Development istry and Life Science, United States Military Academy, West Point, New York, USA • NKT-Cell Functions • Conclusions Jelena S Bezbradica, Institute for Molecular Bioscience, The University of • Acknowledgements Queensland, Brisbane, Queensland, Australia th Luc Van Kaer, Department of Pathology, Microbiology and Immunology, Vander- Online posting date: 15 January 2016 bilt University School of Medicine, Nashville, Tennessee, USA Sebastian Joyce, Veterans Administration Tennessee Valley Healthcare System, Murfreesboro, Tennessee, USA and Department of Pathology, Microbiology and Immunol- ogy, Vanderbilt University School of Medicine, Nashville, Tennessee, USA Based in part on the previous version of this eLS article ‘Natural Killer T Cells’ (2007) by Jelena S Bezbradica, Luc Van Kaer and Sebastian Joyce. Natural killer T (NKT) cells that express the Introduction semi-invariant T-cell receptor are innate-like lymphocytes whose functions are controlled by The immune system, as a constituent of the ten physiologic sys- self and foreign glycolipid ligands. Such ligands tems of the body, has evolved to sense perturbations in the milieu are presented by the antigen-presenting, MHC intérieur (homeostasis) and to actuate an appropriate response class I-like molecule CD1d, which belongs to a so as to restore a set homeostatic state unique to an organism. family of lipid antigen-presenting molecules col- Homeostasis can be disrupted by both internal and external lectively called CD1.