Mechanism of Action of Antidepressants and Mood Stabilizers
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Antinociceptive Effects of Monoamine Reuptake Inhibitors in Assays of Pain-Stimulated and Pain-Depressed Behaviors
Virginia Commonwealth University VCU Scholars Compass Theses and Dissertations Graduate School 2012 Antinociceptive Effects of Monoamine Reuptake Inhibitors in Assays of Pain-Stimulated and Pain-Depressed Behaviors Marisa Rosenberg Virginia Commonwealth University Follow this and additional works at: https://scholarscompass.vcu.edu/etd Part of the Medical Pharmacology Commons © The Author Downloaded from https://scholarscompass.vcu.edu/etd/2715 This Thesis is brought to you for free and open access by the Graduate School at VCU Scholars Compass. It has been accepted for inclusion in Theses and Dissertations by an authorized administrator of VCU Scholars Compass. For more information, please contact [email protected]. ANTINOCICEPTIVE EFFECTS OF MONOAMINE REUPTAKE INHIBITORS IN ASSAYS OF PAIN-STIMULATED AND PAIN-DEPRESSED BEHAVIOR A thesis submitted in partial fulfillment of the requirements for the degree of Master of Science at Virginia Commonwealth University By Marisa B. Rosenberg Bachelor of Science, Temple University, 2008 Advisor: Sidney Stevens Negus, Ph.D. Professor, Department of Pharmacology/Toxicology Virginia Commonwealth University Richmond, VA May, 2012 Acknowledgement First and foremost, I’d like to thank my advisor Dr. Steven Negus, whose unwavering support, guidance and patience throughout my graduate career has helped me become the scientist I am today. His dedication to education, learning and the scientific process has instilled in me a quest for knowledge that I will continue to pursue in life. His thoroughness, attention to detail and understanding of pharmacology has been exemplary to a young person like me just starting out in the field of science. I’d also like to thank all of my committee members (Drs. -

The 5-HT6 Receptor Antagonist SB-271046 Selectively Enhances Excitatory Neurotransmission in the Rat Frontal Cortex and Hippocampus Lee A
The 5-HT6 Receptor Antagonist SB-271046 Selectively Enhances Excitatory Neurotransmission in the Rat Frontal Cortex and Hippocampus Lee A. Dawson, Ph.D., Huy Q. Nguyen, B.S., and Ping Li, B.S. Preclinical evidence has suggested a possible role for the 5-HT6 increases in extracellular glutamate levels in both frontal receptor in the treatment of cognitive dysfunction. However, cortex and dorsal hippocampus, respectively. These effects were currently there is little neurochemical evidence suggesting the completely attenuated by infusion of tetrodotoxin but mechanism(s) which may be involved. Using the selective unaffected by the muscarinic antagonist, atropine. Here we 5-HT6 antagonist SB-271046 and in vivo microdialysis, we demonstrate for the first time the selective enhancement of have evaluated the effects of this compound on the modulation excitatory neurotransmission by SB-271046 in those brain of basal neurotransmitter release within multiple brain regions regions implicated in cognitive and memory function, and of the freely moving rat. SB-271046 produced no change in provide mechanistic evidence in support of a possible basal levels of dopamine (DA), norepinephrine (NE) or 5-HT therapeutic role for 5-HT6 receptor antagonists in the in the striatum, frontal cortex, dorsal hippocampus or nucleus treatment of cognitive and memory dysfunction. accumbens. Similarly, this compound had no effect on [Neuropsychopharmacology 25:662–668, 2001] excitatory neurotransmission in the striatum or nucleus © 2001 American College of Neuropsychopharmacology. accumbens. Conversely, SB-271046 produced 3- and 2-fold Published by Elsevier Science Inc. KEY WORDS: 5-HT6 receptor; SB-271046; Microdialysis; sma et al. 1993; Ruat et al. -

M100907, a Serotonin 5-HT2A Receptor Antagonist and Putative Antipsychotic, Blocks Dizocilpine-Induced Prepulse Inhibition Defic
M100907, a Serotonin 5-HT2A Receptor Antagonist and Putative Antipsychotic, Blocks Dizocilpine-Induced Prepulse Inhibition Deficits in Sprague–Dawley and Wistar Rats Geoffrey B. Varty, Ph.D., Vaishali P. Bakshi, Ph.D., and Mark A. Geyer, Ph.D. a In a recent study using Wistar rats, the serotonergic 5-HT2 1 receptor agonist cirazoline disrupts PPI. As risperidone a receptor antagonists ketanserin and risperidone reduced the and M100907 have affinity at the 1 receptor, a final study disruptive effects of the noncompetitive N-methyl-D- examined whether M100907 would block the effects of aspartate (NMDA) antagonist dizocilpine on prepulse cirazoline on PPI. Risperidone partially, but inhibition (PPI), suggesting that there is an interaction nonsignificantly, reduced the effects of dizocilpine in Wistar between serotonin and glutamate in the modulation of PPI. rats, although this effect was smaller than previously In contrast, studies using the noncompetitive NMDA reported. Consistent with previous studies, risperidone did antagonist phencyclidine (PCP) in Sprague–Dawley rats not alter the effects of dizocilpine in Sprague–Dawley rats. found no effect with 5-HT2 antagonists. To test the hypothesis Most importantly, M100907 pretreatment fully blocked the that strain differences might explain the discrepancy in effect of dizocilpine in both strains; whereas SDZ SER 082 these findings, risperidone was tested for its ability to had no effect. M100907 had no influence on PPI by itself reduce the PPI-disruptive effects of dizocilpine in Wistar and did not reduce the effects of cirazoline on PPI. These and Sprague–Dawley rats. Furthermore, to determine studies confirm the suggestion that serotonin and glutamate which serotonergic receptor subtype may mediate this effect, interact in modulating PPI and indicate that the 5-HT2A the 5-HT2A receptor antagonist M100907 (formerly MDL receptor subtype mediates this interaction. -

Early-Life Experience, Epigenetics, and the Developing Brain
Neuropsychopharmacology REVIEWS (2015) 40, 141–153 & 2015 American College of Neuropsychopharmacology. All rights reserved 0893-133X/15 REVIEW ............................................................................................................................................................... www.neuropsychopharmacologyreviews.org 141 Early-Life Experience, Epigenetics, and the Developing Brain 1 ,1 Marija Kundakovic and Frances A Champagne* 1Department of Psychology, Columbia University, New York, NY, USA Development is a dynamic process that involves interplay between genes and the environment. In mammals, the quality of the postnatal environment is shaped by parent–offspring interactions that promote growth and survival and can lead to divergent developmental trajectories with implications for later-life neurobiological and behavioral characteristics. Emerging evidence suggests that epigenetic factors (ie, DNA methylation, posttranslational histone modifications, and small non- coding RNAs) may have a critical role in these parental care effects. Although this evidence is drawn primarily from rodent studies, there is increasing support for these effects in humans. Through these molecular mechanisms, variation in risk of psychopathology may emerge, particularly as a consequence of early-life neglect and abuse. Here we will highlight evidence of dynamic epigenetic changes in the developing brain in response to variation in the quality of postnatal parent–offspring interactions. The recruitment of epigenetic pathways for the -

Guidance on the Use of Mood Stabilizers for the Treatment of Bipolar Affective Disorder Version 2
Guidance on the use of mood stabilizers for the treatment of bipolar affective disorder Version 2 RATIFYING COMMITTEE DRUGS AND THERAPEUTICS GROUP DATE RATIFIED July 2015 REPLACES Version 1 dated July 2013 NEXT REVIEW DATE July 2017 POLICY AUTHORS Jules Haste, Lead Pharmacist, Brighton and Hove Members of the Pharmacy Team (contributors are listed overleaf) If you require this document in an alternative format, i.e. easy read, large text, audio or Braille please contact the pharmacy team on 01243 623349 Page 1 of 49 Contributors Jed Hewitt, Chief Pharmacist - Governance & Professional Practice James Atkinson, Pharmacist Team Leader Mental Health and Community Services Miguel Gomez, Lead Pharmacist, Worthing. Hilary Garforth, Lead Pharmacist, Chichester. Pauline Daw, Lead Pharmacist (CRHTs & AOT), East Sussex. Iftekhar Khan, Lead Pharmacist (S&F Service), East Sussex. Graham Brown, Lead Pharmacist CAMHS & EIS. Gus Fernandez, Specialist Pharmacist MI and MH Lisa Stanton, Specialist Pharmacist Early Intervention Services & Learning Disabilities. Nana Tomova, Specialist Pharmacist, Crawley. Page 2 of 49 Section Title Page Number Introduction and Key Points 4 1. General principles in the treatment of acute mania 6 2. General principles in the treatment of bipolar depression 8 3. General principles in long term treatment 10 4. Rapid cycling 13 5. Physical health 13 6. Treatment in special situations 6.1 Pregnancy 15 6.2 Breast-feeding 17 6.3 Older adults 19 6.4 Children and adolescents 22 6.5 Learning disabilities 29 6.6 Cardiac dysfunction 30 6.7 Renal dysfunction 34 6.8 Hepatic dysfunction 37 6.9 Epilepsy 41 7. The risk of switching to mania with antidepressants 43 8. -

Homicide and Associated Steroid Acute Psychosis: a Case Report
Hindawi Publishing Corporation Case Reports in Medicine Volume 2011, Article ID 564521, 4 pages doi:10.1155/2011/564521 Case Report Homicide and Associated Steroid Acute Psychosis: A Case Report G. Airagnes,1, 2 C. Rouge-Maillart,1, 3 J.-B. Garre,2, 3 and B. Gohier2 1 Service de M´edecine L´egale, CHU d’Angers, 49933 Angers Cedex 09, France 2 D´epartement de Psychiatrie et de Psychologie m´edicale, CHU d’Angers, 49933 Angers Cedex 09, France 3 IFR 132, Universit´e d’Angers, 49035 Angers, France Correspondence should be addressed to G. Airagnes, [email protected] Received 22 June 2011; Revised 26 August 2011; Accepted 26 September 2011 Academic Editor: Massimo Gallerani Copyright © 2011 G. Airagnes et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. We report the case of an old man treated with methylprednisolone for chronic lymphoid leukemia. After two months of treatment, he declared an acute steroid psychosis and beat his wife to death. Steroids were stopped and the psychotic symptoms subsided, but his condition declined very quickly. The clinical course was complicated by a major depressive disorder with suicidal ideas, due to the steroid stoppage, the leukemia progressed, and by a sudden onset of a fatal pulmonary embolism. This clinical case highlights the importance of early detection of steroid psychosis and proposes, should treatment not be stopped, a strategy of dose reduction combined with a mood stabilizer or antipsychotic treatment. -

Appendix D. Study Characteristics
Appendix D. Study Characteristics Table D1. Study characteristics Participant Author Study Study Characteristics Treatment Characteristics Outcomes Reported Characteristics Conclusions Alacqua et al., Recruitment dates: Enrolled: 73 Treatment duration: 3 mo Benefits: NR Adverse events 2008 96 Jan 2002 to Dec 2003 Analyzed: 73 Run-in phase: No occurred frequently Completed: 50 Run-in phase duration: NR Harms: Behavioral during first 3 months Country: Italy Study design: issues, dyskinesia, of treatment with Retrospective cohort GROUP 1 Permitted drugs: NR dystonia, atypical Condition N: 2 dermatologic AE, liver antipsychotics. category: Mixed Diagnostic criteria: Age, mean±SD (range): Prohibited drugs: NR function, hepatic conditions (ADHD, DSM-IV 15.5±0.7 volume, prolactin, ASD, Males %: 50 GROUP 1 prolactin-related AE, schizophrenia- Setting: Caucasian %: NR Drug name: Clozapine sedation, sleepness, related, tics) Outpatient/community Diagnostic breakdown Dosing variability: variable total AE, weight (n): psychosis (1), Target dose (mg/day): NR change Funding: NR Inclusion criteria: (1) schizophrenia (1) Daily dose (mg/day), mean±SD ≤18 yr, (2) received an Treatment naïve (n): all (range): 150±70.1 Newcastle-Ottawa incident treatment with Inpatients (n): NR Concurrent treatments: NR Scale: 6/8 stars atypical antipsychotics First episode psychosis or SSRIs during the (n): NR GROUP 2 study period Comorbidities: NR Drug name: Olanzapine Dosing variability: variable Exclusion criteria: GROUP 2 Target dose (mg/day): NR NR N: 24 Daily dose -

Neuropsychopharmacology (2015) 40, 2853–2855 © 2015 American College of Neuropsychopharmacology
Neuropsychopharmacology (2015) 40, 2853–2855 © 2015 American College of Neuropsychopharmacology. All rights reserved 0893-133X/15 www.neuropsychopharmacology.org Commentary Neuropsychopharmacology: Reflections on 40 Volumes Alan Frazer*,1 1 Department of Pharmacology, University of Texas Health Science Center at San Antonio, San Antonio, Texas, USA Neuropsychopharmacology (2015) 40, 2853–2855; doi:10.1038/npp.2015.290 This issue completes volume 40 of Neuropsychopharmaco- methodology has not followed in concert with pre-clinical logy (NPP), with the first issue published in December, 1987. advances such that most efficacy studies still lump together This should be distinguished from our 40th anniversary; what are heterogeneous groups of patients. One might say for the first several years, issues were thin, months were how can we subdivide in the absence of biomarkers, which skipped, and multiple volumes were published per year. To do not exist? But one might also ask how can we hope to find commemorate this accomplishment, the editor-in-chief a biomarker among a group of patients that might have 3, 7, (Dr Carlezon) asked me as in-coming ACNP President to or 10 different pathologies, all leading to the same behavioral write a brief overview of my views on the developments in output. Talk about the chicken and the egg! our field over this time period, using highly cited articles Another theme that continues into the present is the time- published in NPP as a guide. dependent, downstream consequences of acute actions of Early on, two articles highlight themes that continue today. some type of psychotherapeutic drugs that correlate better One is the development of animal models for psychiatric with optimal clinical improvement. -

Pharmacogenomics: the Promise of Personalized Medicine for CNS Disorders
Neuropsychopharmacology REVIEWS (2009) 34, 159–172 & 2009 Nature Publishing Group All rights reserved 0893-133X/09 $30.00 REVIEW ............................................................................................................................................................... www.neuropsychopharmacology.org 159 Pharmacogenomics: The Promise of Personalized Medicine for CNS Disorders Jose de Leon*,1,2,3,4 1 2 Mental Health Research Center, Eastern State Hospital, University of Kentucky, Lexington, KY, USA; College of Medicine, 3 4 University of Kentucky, Lexington, KY, USA; College of Pharmacy, University of Kentucky, Lexington, KY, USA; Psychiatry and Neurosciences Research Group (CTS-549), Institute of Neurosciences, Medical School, University of Granada, Granada, Spain This review focuses first on the concept of pharmacogenomics and its related concepts (biomarkers and personalized prescription). Next, the first generation of five DNA pharmacogenomic tests used in the clinical practice of psychiatry is briefly reviewed. Then the possible involvement of these pharmacogenomic tests in the exploration of early clinical proof of mechanism is described by using two of the tests and one example from the pharmaceutical industry (iloperidone clinical trials). The initial attempts to use other microarray tests (measuring RNA expression) as peripheral biomarkers for CNS disorders are briefly described. Then the challenge of taking pharmacogenomic tests (compared to drugs) into clinical practice is explained by focusing on regulatory -

Page 1 of 85 Reference ID: 3091122
HIGHLIGHTS OF PRESCRIBING INFORMATION As an adjunct to 2-5 mg 5-10 mg 15 mg These highlights do not include all the information needed to use ABILIFY antidepressants for the /day /day /day safely and effectively. See full prescribing information for ABILIFY. treatment of major ® ABILIFY (aripiprazole) Tablets depressive disorder – adults ® ABILIFY DISCMELT (aripiprazole) Orally Disintegrating Tablets (2.3) ® Irritability associated with 2 mg/day 5-10 mg/day 15 mg/day ABILIFY (aripiprazole) Oral Solution ® autistic disorder – pediatric ABILIFY (aripiprazole) Injection FOR INTRAMUSCULAR USE ONLY patients (2.4) Initial U.S. Approval: 2002 Agitation associated with 9.75 mg /1.3 30 mg/day schizophrenia or bipolar mL injected injected WARNINGS: INCREASED MORTALITY IN ELDERLY mania – adults (2.5) IM IM PATIENTS WITH DEMENTIA-RELATED PSYCHOSIS and • Oral formulations: Administer once daily without regard to meals (2) SUICIDALITY AND ANTIDEPRESSANT DRUGS • IM injection: Wait at least 2 hours between doses. Maximum daily dose See full prescribing information for complete boxed warning. 30 mg (2.5) • Elderly patients with dementia-related psychosis treated with ----------------------DOSAGE FORMS AND STRENGTHS--------------------- antipsychotic drugs are at an increased risk of death. ABILIFY is not approved for the treatment of patients with dementia-related • Tablets: 2 mg, 5 mg, 10 mg, 15 mg, 20 mg, and 30 mg (3) psychosis. (5.1) • Orally Disintegrating Tablets: 10 mg and 15 mg (3) • Oral Solution: 1 mg/mL (3) • Children, adolescents, and young adults taking antidepressants for • Injection: 9.75 mg/1.3 mL single-dose vial (3) major depressive disorder (MDD) and other psychiatric disorders are at increased risk of suicidal thinking and behavior. -
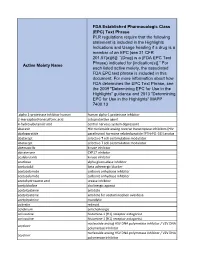
Active Moiety Name FDA Established Pharmacologic Class (EPC) Text
FDA Established Pharmacologic Class (EPC) Text Phrase PLR regulations require that the following statement is included in the Highlights Indications and Usage heading if a drug is a member of an EPC [see 21 CFR 201.57(a)(6)]: “(Drug) is a (FDA EPC Text Phrase) indicated for [indication(s)].” For Active Moiety Name each listed active moiety, the associated FDA EPC text phrase is included in this document. For more information about how FDA determines the EPC Text Phrase, see the 2009 "Determining EPC for Use in the Highlights" guidance and 2013 "Determining EPC for Use in the Highlights" MAPP 7400.13. .alpha. -
5-HT1A Receptor Agonist Affects Fear Conditioning Through Stimulations of the Postsynaptic 5-HT1A Receptors in the Title Hippocampus and Amygdala
5-HT1A receptor agonist affects fear conditioning through stimulations of the postsynaptic 5-HT1A receptors in the Title hippocampus and amygdala Author(s) Li, XiaoBai; Inoue, Takeshi; Abekawa, Tomohiro; Weng, ShiMin; Nakagawa, Shin; Izumi, Takeshi; Koyama, Tsukasa European Journal of Pharmacology, 532(1-2), 74-80 Citation https://doi.org/10.1016/j.ejphar.2005.12.008 Issue Date 2006-02-17 Doc URL http://hdl.handle.net/2115/8408 Type article (author version) File Information EJP22907revisedFinal.pdf Instructions for use Hokkaido University Collection of Scholarly and Academic Papers : HUSCAP 5-HT1A receptor agonist affects fear conditioning through stimulations of the postsynaptic 5-HT1A receptors in the hippocampus and amygdala. XiaoBai Lia, b, Takeshi Inouea, Tomohiro Abekawaa, ShiMin Weng b, Shin Nakagawaa, Takeshi Izumia, Tsukasa Koyamaa a Department of Psychiatry, Neural Function, Hokkaido University Graduate School of Medicine, North 15, West 7, Kita-ku, Sapporo, 060-8638, Japan bShanghai Mental Health Center, 600, Wan Ping Nan Lu, Shanghai 200030, China Correspondence to: Takeshi Inoue, MD, PhD, Department of Psychiatry, Neural Function, Hokkaido University Graduate School of Medicine, North 15, West 7, Kita-ku, Sapporo 060-8638, Japan. Tel: +81-11-706-5160; Fax: +81-11-706-5081; E-mail: [email protected] Abstract Evidence from preclinical and clinical studies has shown that 5-HT1A receptor agonists have anxiolytic actions. The anxiolytic actions of 5-HT1A receptor agonists have been tested by our previous studies using fear conditioning. However, little is known about the brain regions of anxiolytic actions of 5-HT1A receptor agonists in this paradigm. In the present study, we investigated the effects of bilateral microinjections of flesinoxan, a selective 5-HT1A receptor agonist, into the hippocampus, amygdala and medial prefrontal cortex on the expression of contextual conditioned freezing and the defecation induced by conditioned fear stress in rats.