Mitochondrial D-Loop Sequence Variation and Maternal Lineage in the Endangered Cleveland Bay Horse
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Thoroughbred Horses
Thoroughbred Horses Visit Funny Cide at the KHP Hall of Champions! A long time ago, man tamed the horse. People used horses to farm and to ride. Today, people also race horses. The most popular breed for horse racing is the Thoroughbred. The Thoroughbred is the only horse that can compete in the Kentucky Derby. * This educational packet is intended for third, fourth, and fifth graders. It may be complete in small groups or individually. ! Name:_______________________________ Date:________________________________ The Life Cycle of a Thoroughbred Racehorse Racehorses are born on farms. 1 Baby horses are called foals. ! ! Mother horses are called Mares. Foals live with their mothers. Father horses are called Stallions. 2 ! ! When foals are about six months old they are weaned, meaning separated from their mothers. 3 Weanlings live in a herd made of up horses their age. ! ! When horses turn one year old, they are called yearlings. At this point, boy horses are called colts, and girl horses are called fillies. 4 ! ! 5 Horses start racing at two years old. ! ! Racehorses retire on farms after (hopefully) long careers. Some racehorses become pleasure horses, while others are bred to produce more 6 racehorses. ! ! What about horses? Where do horses live? Horses live in barns and outside. In a barn, a horse lives in a stall. Outside, a horse lives in a pasture. ! White Prince A Rare White Thoroughbred Visit him at the KHP! ! ! What do horses eat? Horses eat a lot during the day. From the time they are born, until they are about 5 months old, foals need to drink their mother’s milk. -

Herd/Partnership Number: ______Name: ______
Herd/Partnership Number: ________________ Name: __________________ BEFORE YOU SEND IN YOUR 2017 RARE BREEDS CLAIM, PLEASE USE THE CHECKLIST BELOW TO ENSURE ALL REQUIRED DOCUMENTATION IS ATTACHED TO YOUR CLAIM For Equines Only (Connemara Pony, Irish Draught, Kerry Bog Pony) Declaration form provided, completed and signed. Record Sheet provided, completed with 2017 details and signed. Proof of membership of Breed Society as issued. The original document or a certified true copy stamped as a true copy by a Department of Agriculture office, your local DVO office, an Agricultural Consultant or a Teagasc Advisor. (Connemara Pony Breeders Society, Kerry Bog Pony Co-Operative Society or The Irish Horse Board (Irish Draught Horse Breed only)) The original passport for each pony/horse you are claiming for in 2017 or a certified true copy of passport. Pages required are front page, markings page and ownership page all stamped as a true copy by a Department of Agriculture office, your local DVO office, an Agricultural Consultant or a Teagasc Advisor. Breeding females must produce registered offspring before the end of the contract; documentary evidence of offspring, from the Breed Society, must be provided. For Bovines Only (Kerry Cattle, Dexter, Irish Maol) Declaration form provided, completed and signed. Record Sheet provided completed with 2017 details and signed. Proof of membership of Breed Society for 2017. The original document or a certified true copy stamped as a true copy by a Department of Agriculture office, your local DVO office, an Agricultural Consultant or a Teagasc Advisor. (Kerry Cattle Society Ltd, Irish Moiled Cattle Society or UK Dexter Cattle Society) Birth Certificate(s)/Birth Notification(s) for each bovine you are claiming for. -

Stewardship Awards Sana
STEWARDSHIP AWARDS of NORTH AMERICA – SANA 2008 SANA SPECIAL AWARD - Winners SANA MOST VERSATILE (MV) AWARDS one In-hand class, one Ridden class, and any one discipline class (Over Fences, Driven, or Dressage) SANA Most Versatile Pony (MVP) - MVP Rosette sponsored by the Equus Survival Trust; travel bag Sponsored by Equine Journal Magazine #106 Black Brandy Pride of the Rock (Newfoundland gelding) Colleen Donald & Meridith Jack, ONTARIO SANA Most Versatile Horse (MVH) - MVP Rosette sponsored by the Equus SurvivalTrust; horse blanket Sponsored by Equine Journal Magazine #114 Metman (Akhal-Teke stallion) Anne-Marie Rasch, MI SANA -MOST VERSATILE BREED AWARDS MV SMALL PONY BREEDS -MV Dartmoor: Rosette sponsored by EndangeredEquines.com No ridden qualifiers -MV Exmoor: Rosette sponsored by EndangeredEquines.com #191 Marlyn Domino (gelding) Katie McCaffrey, NY -MV Gotland Pony: Book on Wild Gotlands Sponsored by - Birgitta Cramer / Gotland Breeder's Organization in Gotland, SWEDEN #165 Sundance (gelding) Amanda Wells, KY MV LARGE PONY BREEDS -MV Dales Pony: DPS Rosette - Sponsored by Dales Pony Society (UK) #202 Sowemire Rose (mare) Gayla Driving Center, KY -MV Fell Pony: Rosette - Sponsored by BroughHill Fells (NC) – Rosette #177 Florence (gelding) Dream Hayven Farm/ Melissa Kreuzer, WI -MV Highland Pony: HPS Rosette – Highland Pony Society (Scotland) #103 Rob Roy O’the Glenns (gelding) Judy Brescia, VA -MV Newfoundland Pony: Rosette – Sponsored by Stableways.com #106 Black Brandy Pride of the Rock (gelding) Collen Donald & Meredeth Jack -

Multiple Choice Choose the Answer That Best Completes Each Statement Or Question
Name Date Hour 9 The Horse Industry Multiple Choice Choose the answer that best completes each statement or question. _______ 1. The horse was first domesticated in Europe and Asia about ____ . A. 1,000 years ago B. 3,000 years ago C. 5,000 years ago D. 8,000 years ago _______ 2. Horses were brought to the New World by Spanish explorers in the ____ . A. 15th century B. 16th century C. 17th century D. 18th century _______ 3. Horses are measured in terms of hands with a hand being ____ . A. two inches B. four inches C. six inches D. eight inches _______ 4. Ponies are shorter than horses and can be anywhere from 8 to ____ . A. 10 hands high B. 12.2 hands high C. 14.2 hands high D. 16 hands high _______ 5. Which horse breed is considered the oldest purebred horse in the world? A. Arabian B. Appaloosa C. Thoroughbred D. Quarter Horse Introduction to Agriscience | Unit 9 Test CIMC 1 _______ 6. Which horse breed has as one of its characteristics a distinctive spotted coat? A. Arabian B. Appaloosa C. Thoroughbred D. Quarter Horse _______ 7. Which horse breed was developed in the United States and got its name because of its great speed at short distances? A. Arabian B. Appaloosa C. Thoroughbred D. Quarter Horse _______ 8. Which horse breed was developed in the deserts of the Middle East? A. Arabian B. Appaloosa C. Thoroughbred D. Quarter Horse _______ 9. Which horse breed has a head characterized by a dished profile, prominent eye, large nostrils, and small muzzle? A. -

Irish Breeds Quiz Answers 1. What County In
Irish Breeds Quiz Answers 1. What county in Ireland does the Connemara Pony originate from? County Galway 2. Traditionally, for what purpose was the Irish Cob bred? To pull a cart / wagon To travel long distances To be suitable for children To be strong and sturdy 3. List three typical traits of the Irish Draught: 1. Versatile 2. Intelligent 3. Kind / willing nature Strong and sturdy, pulling / staying power / hardy 4. What are the two main composite breeds of the Irish Sport Horse? 1. Irish Draught 2. Thoroughbred 5. What is the smallest Irish native breed? Kerry Bog Pony 6. What three horse breeds can be included in a horse’s pedigree for it to qualify as a Traditional Irish Horse? Connemara Pony, Irish Draught, Thoroughbred 7. List three typical traits of the Connemara Pony: 1. Sure footed 2. Hardy 3. Calm / willing / kind temperament Staying power / intelligent / sound / athletic / versatile 8. Name one typical conformation trait of the Irish Cob: Stout, powerful, wide / short back, wide chest / good bone 9. For what activities were the Irish Draught originally bred? Farm work / pulling machinery, riding, hunting, driving 10. For what activities are Irish Draughts bred for today? Leisure / riding horses / allrounders, competition, cross breeding 11. What traits make the Irish Sport Horse so well suited to Equestrian sport today? Athleticism, jumping ability, courage, intelligence, soundness, kind temperament 12. What are the two main reasons for producing Kerry Bog Ponies? 1. To pull machinery 2. As riding ponies for children Companion ponies Showing . -

Population Genetic Analysis of the Estonian Native Horse Suggests Diverse and Distinct Genetics, Ancient Origin and Contribution from Unique Patrilines
G C A T T A C G G C A T genes Article Population Genetic Analysis of the Estonian Native Horse Suggests Diverse and Distinct Genetics, Ancient Origin and Contribution from Unique Patrilines Caitlin Castaneda 1 , Rytis Juras 1, Anas Khanshour 2, Ingrid Randlaht 3, Barbara Wallner 4, Doris Rigler 4, Gabriella Lindgren 5,6 , Terje Raudsepp 1,* and E. Gus Cothran 1,* 1 College of Veterinary Medicine and Biomedical Sciences, Texas A&M University, College Station, TX 77843, USA 2 Sarah M. and Charles E. Seay Center for Musculoskeletal Research, Texas Scottish Rite Hospital for Children, Dallas, TX 75219, USA 3 Estonian Native Horse Conservation Society, 93814 Kuressaare, Saaremaa, Estonia 4 Institute of Animal Breeding and Genetics, University of Veterinary Medicine Vienna, 1210 Vienna, Austria 5 Department of Animal Breeding and Genetics, Swedish University of Agricultural Sciences, 75007 Uppsala, Sweden 6 Livestock Genetics, Department of Biosystems, KU Leuven, B-3001 Leuven, Belgium * Correspondence: [email protected] (T.R.); [email protected] (E.G.C.) Received: 9 August 2019; Accepted: 13 August 2019; Published: 20 August 2019 Abstract: The Estonian Native Horse (ENH) is a medium-size pony found mainly in the western islands of Estonia and is well-adapted to the harsh northern climate and poor pastures. The ancestry of the ENH is debated, including alleged claims about direct descendance from the extinct Tarpan. Here we conducted a detailed analysis of the genetic makeup and relationships of the ENH based on the genotypes of 15 autosomal short tandem repeats (STRs), 18 Y chromosomal single nucleotide polymorphisms (SNPs), mitochondrial D-loop sequence and lateral gait allele in DMRT3. -

List of Horse Breeds 1 List of Horse Breeds
List of horse breeds 1 List of horse breeds This page is a list of horse and pony breeds, and also includes terms used to describe types of horse that are not breeds but are commonly mistaken for breeds. While there is no scientifically accepted definition of the term "breed,"[1] a breed is defined generally as having distinct true-breeding characteristics over a number of generations; its members may be called "purebred". In most cases, bloodlines of horse breeds are recorded with a breed registry. However, in horses, the concept is somewhat flexible, as open stud books are created for developing horse breeds that are not yet fully true-breeding. Registries also are considered the authority as to whether a given breed is listed as Light or saddle horse breeds a "horse" or a "pony". There are also a number of "color breed", sport horse, and gaited horse registries for horses with various phenotypes or other traits, which admit any animal fitting a given set of physical characteristics, even if there is little or no evidence of the trait being a true-breeding characteristic. Other recording entities or specialty organizations may recognize horses from multiple breeds, thus, for the purposes of this article, such animals are classified as a "type" rather than a "breed". The breeds and types listed here are those that already have a Wikipedia article. For a more extensive list, see the List of all horse breeds in DAD-IS. Heavy or draft horse breeds For additional information, see horse breed, horse breeding and the individual articles listed below. -

New Forest & Hampshire County Show 2019 Results
New Forest & Hampshire County Show 2019 Results EQUINE SECTION ARAB IN HAND - TUESDAY H1 - PURE ARAB - Filly, colt or gelding, yearling, two or three year olds, any height Prize Catalogue Exhibitor Name Animal Name Number 1st 2 ZEBEDEE, MRS STEPHANIE EASTWORTH BERNINI 2nd 1 MARSH, MISS JOANNE FOREVER DISTINCT EQUINE SECTION ARAB IN HAND - TUESDAY H2 - PURE ARAB - Mare, stallion or gelding, four years old and over, any height Prize Catalogue Exhibitor Name Animal Name Number 1st 5 SMALLEY, MISS SUSAN BC IBN HEJAZ EQUINE SECTION ANGLO AND PART BRED ARAB IN HAND - TUESDAY H3 - ARAB, ANGLO AND PART BRED ARAB - Filly, colt or gelding, yearling, two or three year olds, any height Prize Catalogue Exhibitor Name Animal Name Number 1st 9 HARRIS, KIRSTIE TIGER MOON 2nd 11 CHATLEY, WILLIAM NEWFIELDEN NEW BEGINNINGS 3rd 10 BELL, MS DEBBIE DOWHILLS BEAU SOLEIL EQUINE SECTION ANGLO AND PART BRED ARAB IN HAND - TUESDAY H4 - ARAB, ANGLO AND PART BRED ARAB - Mare, stallion or gelding, four years old and over, any height Prize Catalogue Exhibitor Name Animal Name Number 1st 17 GREGORY, MRS STEPHANIE LITTLETONS ADDICTION 2nd 19 GOOD, MISS KATIE RENDENE GYPSY CHARM 3rd 12 MCKELL, MRS LISA CHIDDOCK CURFEW 4th 15 HARDING, MISS MARTINE MAGNIKS LAST EDITION 5th 18 KNOCK, MRS K HAMMONDS HALF TIME 6th 16 BOOTH, MR & MRS ANTHONY LOWLAND PRETAPORTER New Forest & Hampshire County Show 2019 Results EQUINE SECTION COMPETITION PONY IN HAND - TUESDAY H5 - IN HAND COMPETITION PONY - One, two and three year olds, colt, filly or gelding, not exceeding 153cms Prize Catalogue -

Putting the Cart Before the Horse: Barriers to Enforcing a Code of Ethics for Thoroughbred Auctions in the United States Catharine Altier
Brooklyn Law Review Volume 72 | Issue 3 Article 4 2007 Putting the Cart Before the Horse: Barriers to Enforcing a Code of Ethics for Thoroughbred Auctions in the United States Catharine Altier Follow this and additional works at: https://brooklynworks.brooklaw.edu/blr Recommended Citation Catharine Altier, Putting the Cart Before the Horse: Barriers to Enforcing a Code of Ethics for Thoroughbred Auctions in the United States, 72 Brook. L. Rev. (2007). Available at: https://brooklynworks.brooklaw.edu/blr/vol72/iss3/4 This Note is brought to you for free and open access by the Law Journals at BrooklynWorks. It has been accepted for inclusion in Brooklyn Law Review by an authorized editor of BrooklynWorks. NOTES Putting the Cart Before the Horse BARRIERS TO ENFORCING A CODE OF ETHICS FOR THOROUGHBRED AUCTIONS IN THE UNITED STATES I. INTRODUCTION In today’s thoroughbred racing world, sometimes what you see is not at all what you get. Even veteran horsemen will admit that “[t]here is a fine line between the showmanship of showing a horse at its fullest and fraud.”1 Most surprisingly, this deception often begins long before a thoroughbred has even run its first race. Sales practices that may appear fraudulent to horse racing outsiders are tolerated, or even accepted as customary practice, at thoroughbred auctions.2 For example, before being sold, horses are sometimes injected with steroids to make their chests appear stronger.3 Agents, hired to bid for prospective owners, have been caught defrauding their principals by colluding with sellers and accepting undisclosed commissions.4 Sellers even use agents to bid on their own 1 Joe Drape, No Gift Horses Here, So Look in Their Mouths, N.Y. -
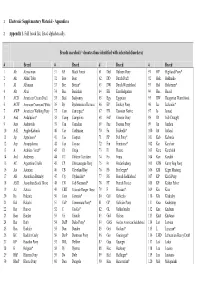
Electronic Supplementary Material - Appendices
1 Electronic Supplementary Material - Appendices 2 Appendix 1. Full breed list, listed alphabetically. Breeds searched (* denotes those identified with inherited disorders) # Breed # Breed # Breed # Breed 1 Ab Abyssinian 31 BF Black Forest 61 Dul Dülmen Pony 91 HP Highland Pony* 2 Ak Akhal Teke 32 Boe Boer 62 DD Dutch Draft 92 Hok Hokkaido 3 Al Albanian 33 Bre Breton* 63 DW Dutch Warmblood 93 Hol Holsteiner* 4 Alt Altai 34 Buc Buckskin 64 EB East Bulgarian 94 Huc Hucul 5 ACD American Cream Draft 35 Bud Budyonny 65 Egy Egyptian 95 HW Hungarian Warmblood 6 ACW American Creme and White 36 By Byelorussian Harness 66 EP Eriskay Pony 96 Ice Icelandic* 7 AWP American Walking Pony 37 Cam Camargue* 67 EN Estonian Native 97 Io Iomud 8 And Andalusian* 38 Camp Campolina 68 ExP Exmoor Pony 98 ID Irish Draught 9 Anv Andravida 39 Can Canadian 69 Fae Faeroes Pony 99 Jin Jinzhou 10 A-K Anglo-Kabarda 40 Car Carthusian 70 Fa Falabella* 100 Jut Jutland 11 Ap Appaloosa* 41 Cas Caspian 71 FP Fell Pony* 101 Kab Kabarda 12 Arp Araappaloosa 42 Cay Cayuse 72 Fin Finnhorse* 102 Kar Karabair 13 A Arabian / Arab* 43 Ch Cheju 73 Fl Fleuve 103 Kara Karabakh 14 Ard Ardennes 44 CC Chilean Corralero 74 Fo Fouta 104 Kaz Kazakh 15 AC Argentine Criollo 45 CP Chincoteague Pony 75 Fr Frederiksborg 105 KPB Kerry Bog Pony 16 Ast Asturian 46 CB Cleveland Bay 76 Fb Freiberger* 106 KM Kiger Mustang 17 AB Australian Brumby 47 Cly Clydesdale* 77 FS French Saddlebred 107 KP Kirdi Pony 18 ASH Australian Stock Horse 48 CN Cob Normand* 78 FT French Trotter 108 KF Kisber Felver 19 Az Azteca -

Discriminant Canonical Analysis of the Contribution of Spanish and Arabian Purebred Horses to the Genetic Diversity and Population Structure of Hispano-Arabian Horses
UC Davis UC Davis Previously Published Works Title Discriminant Canonical Analysis of the Contribution of Spanish and Arabian Purebred Horses to the Genetic Diversity and Population Structure of Hispano-Arabian Horses. Permalink https://escholarship.org/uc/item/8w77w522 Journal Animals : an open access journal from MDPI, 11(2) ISSN 2076-2615 Authors Marín Navas, Carmen Delgado Bermejo, Juan Vicente McLean, Amy Katherine et al. Publication Date 2021-01-21 DOI 10.3390/ani11020269 Peer reviewed eScholarship.org Powered by the California Digital Library University of California animals Article Discriminant Canonical Analysis of the Contribution of Spanish and Arabian Purebred Horses to the Genetic Diversity and Population Structure of Hispano-Arabian Horses Carmen Marín Navas 1 , Juan Vicente Delgado Bermejo 1 , Amy Katherine McLean 2 , José Manuel León Jurado 3, Antonio Rodriguez de la Borbolla y Ruiberriz de Torres 4 and Francisco Javier Navas González 1,* 1 Department of Genetics, Faculty of Veterinary Sciences, University of Córdoba, 14071 Córdoba, Spain; [email protected] (C.M.N.); [email protected] (J.V.D.B.) 2 Department of Animal Science, University of California Davis, Davis, CA 95617, USA; [email protected] 3 Centro Agropecuario Provincial de Córdoba, Diputación Provincial de Córdoba, 14071 Córdoba, Spain; [email protected] 4 Unión Española de Ganaderos de Pura Raza Hispano-Árabe, 41001 Sevilla, Spain; [email protected] * Correspondence: [email protected]; Tel.: +34-957-21-87-06 Simple Summary: The demographic and genetic diversity structure and the contributions of Spanish (PRE) and Arabian Purebred (PRá) horses to the process of conformation of the Hispano-Arabian Citation: Marín Navas, C.; Delgado (Há) horse breed were evaluated. -

The Kerry Bog Pony Brochure
The Kerry Bog Pony www.kerrybogpony.ie Photo: Bob Langrish THE KERRY BOG PONY The Kerry Bog Pony is a small mountain and moorland type pony. The breed originated in Kerry but it is now found all over Ireland. The numbers however are very low and the breed is still critically ▲ Photo: Liz Sugar endangered. It has a fine intelligent head with large kind eyes. It has a strong and well set on neck with a rounded shoulder and compact body. The pony is clean legged with very little feather to its heels. It has good bone, with short cannon bones, short pasterns and upright hooves. The Kerry Bog Pony is extremely hardy, resistant to many equine diseases with great powers of endurance. Its temperament and versatility make it an excellent children’s pony and it can be used ▲ Photo: Gay Keogh by adults for carriage driving and as a pack animal. Though an ancient breed it was only officially recognised by the Department of Agriculture and the European Union in 2004. Photo: Pascal Lando BREED STANDARD SIZE: This is a small pony evolved as such because of its use as a draught animal in the bogs of Kerry over the centuries. Thus, the height of the Kerry Bog pony is 102 cms - 117 cms for Stallions and Geldings and 102 cms - 112cms for Mares. COLOUR: Any strong whole colour is to be found, but colour is generally brown or brownish black and bay. Some chestnut, grey and dun colours are also to be found. COAT: The coat of the Kerry Bog Pony is long and dense and easily capable of withstanding harsh winter conditions without shelter.