A Dual Effect of Ursolic Acid to the Treatment of Multiple Sclerosis Through Both Immunomodulation and Direct Remyelination
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Involvement of CRH Receptors in the Neuroprotective Action of R-Apomorphine in the Striatal 6-OHDA Rat Model
Neuroscience & Medicine, 2013, 4, 299-318 299 Published Online December 2013 (http://www.scirp.org/journal/nm) http://dx.doi.org/10.4236/nm.2013.44044 Involvement of CRH Receptors in the Neuroprotective Action of R-Apomorphine in the Striatal 6-OHDA Rat Model Mustafa Varçin1, Eduard Bentea1, Steven Roosens1, Yvette Michotte1, Sophie Sarre1,2* 1Department of Pharmaceutical Chemistry and Drug Analysis, Center for Neurosciences, Vrije Universiteit Brussel, Faculty of Medicine and Pharmacy, Brussels, Belgium; 2Belgian Pharmacists Association, Medicines Control Laboratory, Brussels, Belgium. Email: *[email protected] Received October 21st, 2013; revised November 20th, 2013; accepted December 10th, 2013 Copyright © 2013 Mustafa Varçin et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. ABSTRACT The dopamine D1-D2 receptor agonist, R-apomorphine, has been shown to be neuroprotective in different models of Parkinson’s disease. Different mechanisms of action for this effect have been proposed, but not verified in the striatal 6-hydroxydopamine rat model. In this study, the expression of a set of genes involved in 1) signaling, 2) growth and differentiation, 3) neuronal regeneration and survival, 4) apoptosis and 5) inflammation in the striatum was measured after a subchronic R-apomorphine treatment (10 mg/kg/day, subcutaneously, during 11 days) in the striatal 6-hydroxy- dopamine rat model. The expression of 84 genes was analysed by using the rat neurotrophins and receptors RT2 Pro- filer™ PCR array. The neuroprotective effects of R-apomorphine in the striatal 6-hydroxydopamine model were con- firmed by neurochemical and behavioural analysis. -

A Bioinformatics Model of Human Diseases on the Basis Of
SUPPLEMENTARY MATERIALS A Bioinformatics Model of Human Diseases on the basis of Differentially Expressed Genes (of Domestic versus Wild Animals) That Are Orthologs of Human Genes Associated with Reproductive-Potential Changes Vasiliev1,2 G, Chadaeva2 I, Rasskazov2 D, Ponomarenko2 P, Sharypova2 E, Drachkova2 I, Bogomolov2 A, Savinkova2 L, Ponomarenko2,* M, Kolchanov2 N, Osadchuk2 A, Oshchepkov2 D, Osadchuk2 L 1 Novosibirsk State University, Novosibirsk 630090, Russia; 2 Institute of Cytology and Genetics, Siberian Branch of Russian Academy of Sciences, Novosibirsk 630090, Russia; * Correspondence: [email protected]. Tel.: +7 (383) 363-4963 ext. 1311 (M.P.) Supplementary data on effects of the human gene underexpression or overexpression under this study on the reproductive potential Table S1. Effects of underexpression or overexpression of the human genes under this study on the reproductive potential according to our estimates [1-5]. ↓ ↑ Human Deficit ( ) Excess ( ) # Gene NSNP Effect on reproductive potential [Reference] ♂♀ NSNP Effect on reproductive potential [Reference] ♂♀ 1 increased risks of preeclampsia as one of the most challenging 1 ACKR1 ← increased risk of atherosclerosis and other coronary artery disease [9] ← [3] problems of modern obstetrics [8] 1 within a model of human diseases using Adcyap1-knockout mice, 3 in a model of human health using transgenic mice overexpressing 2 ADCYAP1 ← → [4] decreased fertility [10] [4] Adcyap1 within only pancreatic β-cells, ameliorated diabetes [11] 2 within a model of human diseases -

Injury-Induced Upregulation of Bfgf and CNTF Mrnas in the Rat Retina
The Journal of Neuroscience, November 1995, 75(11): 7377-7385 Injury-Induced Upregulation of bFGF and CNTF mRNAS in the Rat Retina Rong Wen,1,2 Ying Song,1,2 Tong Cheng,ls2 Michael T. Matthes,2,3 Douglas Yasumura,2’3 Matthew M. LaVail;,s and Roy H. Steinbergi,2 Departments of ‘Physiology, 20phthalmology, and 3Anatomy, University of California, San Francisco, San Francisco, California 94143 Focal mechanical injury to the retina has been shown to sertion without injection, protects photoreceptors near the wound slow or prevent photoreceptor degeneration near the le- (Faktorovich et al., 1990; Silverman and Hughes, 1990; Blair et sion site in two animal models of retinal degeneration, in- al., 1991; Li et al., 1991). In constant light-induced photorecep- herited retinal dystrophy in the Royal College of Surgeons tor degeneration, a similar injury-dependent protection is ob- (RCS) and light damage in albino rats. Thus, when injured, served (Faktorovich et al., 1992). These findings imply that the the rat retina activates a self-protective mechanism to min- retina has a self-protective mechanism whose activation protects imize damage. To identify injury responsive factors and photoreceptors from damage and death. A similar self-protective cells, we examined the mFlNAs for the following factors mechanism appears to exist for retinal ganglion cells (Mansour- and some of their receptors: basic and acidic fibroblast Robey et al., 1994). growth factors (bFGF, aFGF) and FGF receptor-l (FGFRl); Several lines of evidence suggest that endogenous factors that ciliary neurotrophic factor (CNTF) and CNTF receptor (Y promote photoreceptor survival may be involved in this self- (CNTFRa); brain-derived neurotrophic factor (BDNF) and protective mechanism. -

A Growth Factor from Neuronal Cell Lines Stimulates Myelin Protein Synthesis in Mammalian Brain
The Journal of Neuroscience, February 1991, 7 7(2): 327-336 A Growth Factor from Neuronal Cell Lines Stimulates Myelin Protein Synthesis in Mammalian Brain Dana Giulian,’ Beena Johnson,’ Joseph F. Krebs,’ Mischca J. Tapscott,’ and Sandra Honda* Departments of ‘Neurology and *Cell Biology, Baylor College of Medicine, Houston, Texas 77030 Oligodendroglia growth factor (OGF) is a 16-kDa soluble ferentiation of oligodendroglia, as well as many other cell types protein produced by neuronal cell lines. This factor, when (McMorris et al., 1986; Han et al., 1987). The cytokine inter- incubated with brain glia in culture, selectively stimulates leukin-2 (IL-2) has been described by different laboratories as growth of oligodendroglia, the myelin-producing cells of the a growth promoter (Benveniste and Merrill, 1986) as a growth CNS. OGF infused into the cerebral cortex of the adult rat inhibitor (Saneto et al., 1986) or as having no effect upon oli- accelerates the production of myelin proteins as shown by godendroglia (Giulian and Lachman, 1985; Yong et al., 1988). increased specific activity of the myelin enzyme 2’,3’-cyclic All studies of factor effects upon oligodendroglia have been nucleotide 3’-phosphohydrolase (2’,3’-CNPase), by stimu- limited to cell-culture bioassaysand thus await in vivo con- lated synthesis of myelin basic protein, and by elevations in firmation. When attempting to characterize biologic actions of levels of myelin proteolipid protein RNA. The ability of OGF a growth factor, there is a needto usehighly stringent conditions to induce myelin protein production in viva suggests that with enriched cell populations and chemically defined culture neuron-secreted growth factors help to regulate myelin for- medium. -

Original Article the Analysis and Comparison Between Glia Maturation Factor Beta and Gamma Mrna and Protein Expression in Rat Brain and Cells
Int J Clin Exp Med 2016;9(11):21450-21457 www.ijcem.com /ISSN:1940-5901/IJCEM0026827 Original Article The analysis and comparison between glia maturation factor beta and gamma mRNA and protein expression in rat brain and cells Zhaohu Yuan1, Dandan Chen2, Yaming Wei1*, Bingyi Wu3* Departments of 1Blood Transfusion, 2Radiology, Guangzhou First People’s Hospital, Guangzhou Medical Uni- versity, Guangzhou 510180, China; 3Research Center of Clinical Medicine, Nanfang Hospital, Southern Medical University, Guangzhou 510515, China. *Equal contributors. Received February 27, 2016; Accepted August 12, 2016; Epub November 15, 2016; Published November 30, 2016 Abstract: Glia maturation factor, a 17-kDa protein made up of 142 amino acid residues, mainly includes glia matura- tion factor-β (GMFB) and glia maturation factor-γ (GMFG). GMFB plays an important role in the growth and differenti- ation in neuron as well as glia. GMFG, a novel member of the glia maturation factor gene family, locates in the blood and immune system retaining proliferative and differentiated potential. But to date, the distribution of expression and overall function of GMFG in brain remains elusive. For the first time, we proved the existence of GMFG in the central nervous system in the level of gene, protein and cell by Real-time PCR, Western blotting and Immunofluores- cence. In astrocytes, GMFB expressed in all cells and mainly located in cell nucleus, while GMFG only expressed in a part of astrocytes and located in cell nucleus and cytoplasm presenting stellated. In neuron, GMFB located in the whole cell and it is very obviously in axon and dendritic, but the expression of GMFG in neurons is weak and couldn’t be detected in axon and dendritic. -

Brain Derived Neurotrophic Factor (BDNF) in Cardiometabolic Physiology and Diseases
684 Review Derleme Brain derived neurotrophic factor (BDNF) in cardiometabolic physiology and diseases Kardiyometabolik fizyoloji ve hastalıklarda beyinden türemiş nörotrofik faktör (BDNF) İlker Taşçı, Hasan Kutsi Kabul*, Aydoğan Aydoğdu** From Departments of Internal Medicine, *Cardiology and **Endocrinology and Metabolism, Gülhane School of Medicine, Ankara-Turkey ABSTRACT Important advances in our understanding of the relationships between adipose tissue derived peptides, namely adipokines, and their effects on cardiovascular functions have been achieved in recent years. Growing knowledge of adipokine biology is revealing the complexity of these proteins. Adipose tissue releases some other proteins called neurotrophins that are mainly active in central and peripheral nervous system. However, secre- tion and activity of these hormones are not only limited to neuronal cells and tissues, but they also take part in adipose tissue development, energy metabolism, glucose utilization, insulin sensitivity, inflammation, lipoprotein synthesis, and atherosclerosis. In this review, we describe the most recent advances in the functions of brain derived nerve growth factor (BDNF), a major type of neurotrophins, focusing primarily on cardiovascular and metabolic diseases. (Anadolu Kardiyol Derg 2012; 12: 684-8) Key words: Brain derived nerve growth factor, cardiovascular disease, atherosclerosis, dyslipidemia, diabetes mellitus ÖZET Son yıllarda, adipokinler olarak bilinen adipoz doku kaynaklı peptidler ve bunların kardiyovasküler fonksiyonlar üzerine -
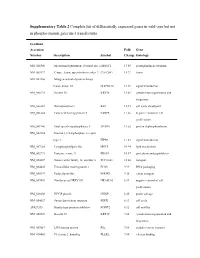
Supplementary Table 2 Complete List of Differentially Expressed Genes in Wild-Type but Not in Phospho-Mutant Galectin-3 Transfectants
Supplementary Table 2 Complete list of differentially expressed genes in wild-type but not in phospho-mutant galectin-3 transfectants GenBank Accession Fold Gene Number Description Symbol Change Ontology NM_020300 Microsomal glutathione S-transferase 1 MGST1 17.65 prostaglandin metabolism NM_005127 C-type lectin, superfamily member 2 CLECSF2 15.22 lectin NM_002446 Mitogen-activated protein kinase kinase kinase 10 MAP3K10 14.39 signal transduction NM_000224 Keratin 18 KRT18 13.61 cytoskeleton organization and biogenesis NM_000321 Retinoblastoma 1 RB1 12.93 cell cycle checkpoint NM_001444 Fatty acid binding protein 5 FABP5 11.66 negative control of cell proliferation NM_001946 Dual specificity phosphatase 6 DUSP6 11.52 protein dephosphorylation NM_002224 Inositol 1,4,5-triphosphate receptor, type 3 ITPR3 11.43 signal transduction NM_007283 Lysophospholipase-like MGLL 10.94 lipid metabolism NM_002771 Protease, serine, 3 PRSS3 10.57 proteolysis and peptidolysis NM_004207 Solute carrier family 16, member 3 SLC16A3 10.04 transport NM_004425 Extracellular matrix protein 1 ECM1 9.99 DNA packaging NM_005397 Podocalyxin-like PODXL 9.35 cation transport NM_007069 Similar to rat HREV107 HRASLS3 8.99 negative control of cell proliferation NM_020200 HHGP protein HHGP 8.25 purine salvage NM_004657 Serum deprivation response SDPR 8.13 cell cycle AF027205 Kunitz-type protease inhibitor SPINT2 8.12 cell motility NM_002276 Keratin 19 KRT19 7.88 cytoskeleton organization and biogenesis NM_003687 LIM domain protein RIL 7.68 oxidative stress response NM_016445 -

Nine Items. the Score Ranges from 1 = Not True to 7 = Absolutely True
Supplementary Material. Fatigue self-assessment scales Fatigue Severity Scale (FSS) [16] Nine items. The score ranges from 1 = not true to 7 = absolutely true. The overall score is given as the mean of the nine items (range 1 to 7). The higher the score is, the more severe is the fatigue. 1. My motivation is lower when I am fatigued. 2. Exercise brings on my fatigue. 3. I am easily fatigued. 4. Fatigue interferes with my physical functioning. 5. Fatigue causes frequent problems to me. 6. My fatigue prevents sustained physical functioning. 7. Fatigue interferes with carrying out certain duties and responsibilities. 8. Fatigue is among my three most disabling symptoms. 9. Fatigue interferes with my work, my family, or social life. MS-Fatigue Severity Scale (MS-FSS) [17] Six items. The score ranges from 1 = not true to 7 = absolutely true. The overall score is given as the mean of the six items (range 1 to 7). The higher the score is, the more severe is the fatigue. 1. Heat worsens my fatigue. 2. Long periods of inactivity worsen the fatigue. 3. Stress worsens the fatigue. 4. Depressive mood worsens the fatigue. 5. Cool temperatures improve my fatigue. 6. Positive experiences improve my fatigue. Modified Fatigue Impact Scale (MFIS) [18] Twenty-one items assessing physical, cognitive, social and total fatigue. The overall score is given as the sum of the responses to 21 single questions (0 = never; 1 = rarely; 2 = sometimes; 3 = frequently; 4 =always; range 0 to 84). The higher the score is, the higher is the impact of fatigue on daily activities. -
A Novel Factor in Olfactory Ensheathing Cell-Astrocyte Crosstalk: Anti-Inflammatory Protein Α-Crystallin B
bioRxiv preprint doi: https://doi.org/10.1101/2020.08.28.273003; this version posted August 28, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. A Novel Factor in Olfactory Ensheathing Cell-Astrocyte Crosstalk: Anti-Inflammatory Protein α- Crystallin B Aybike Saglam1,2*, Anne L. Calof3# and Susan Wray1†‡ 1Cellular & Developmental Neurobiology Section, NINDS, NIH, Bethesda, MD 2Program in Neuroscience & Cognitive Science, Univ. of Maryland, College Park 3Dept of Anatomy & Neurobiology and the Center for Complex Biological Systems, Univ. of California, Irvine, California Key words: astrocyte, neuroinflammation, neurotoxicity, olfactory ensheathing cells (OECs), alpha- crystallin B (CryAB), exosome, nuclear factor kappa B (NFκB) *E-mail: [email protected] #Email: [email protected] †Corresponding author. ‡E-mail: [email protected] Main Points: • Astrocytes uptake OEC-secreted exosomes. • WT OEC-exosomes, but not CryAB-null OEC-exosomes, block nuclear NFκB translocation in astrocytes. • CryAB, and other factors secreted by OECs, suppresses multiple neurotoxicity-associated astrocyte transcripts. Author contributions: AS and SW designed the research. AS performed the experiments. AS analyzed the data and AS, AC and SW wrote the manuscript. bioRxiv preprint doi: https://doi.org/10.1101/2020.08.28.273003; this version posted August 28, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -
Ikb Kinase 2 Determines Oligodendrocyte Loss by Non-Cell-Autonomous Activation of NF-Kb in the Central Nervous System
doi:10.1093/brain/awq359 Brain 2011: 134; 1184–1198 | 1184 BRAIN A JOURNAL OF NEUROLOGY IkB kinase 2 determines oligodendrocyte loss by non-cell-autonomous activation of NF-kB in the central nervous system Jenni Raasch,1,2,* Nicolas Zeller,1,* Geert van Loo,3,4,* Doron Merkler,2,5,* Alexander Mildner,1 Daniel Erny,1 Klaus-Peter Knobeloch,1 John R. Bethea,6 Ari Waisman,7 Markus Knust,1 Domenico Del Turco,8 Thomas Deller,8 Thomas Blank,1 Josef Priller,9 Wolfgang Bru¨ ck,2 Manolis Pasparakis3,10,* and Marco Prinz1,* 1 Department of Neuropathology, University of Freiburg, Breisacher Str. 64, D-79106 Freiburg, Germany 2 Department of Neuropathology, Universita¨ tsmedizin Go¨ ttingen, Robert-Koch-Str. 40, D-37099 Go¨ ttingen, Germany 3 EMBL Mouse Biology Programme, I-00016 Monterotondo, Italy 4 Department of Molecular Biomedical research, VIB and Ghent University, Technologiepark 927, B-9052 Ghent, Belgium 5 Department of Pathology and Immunology, Division of Clincial Pathology, Geneva University Hospital and University of Geneva, Rue Michel-Servet 1, 1211 Geneva, Switzerland 6 The Miami Project to Cure Paralysis, Miller School of Medicine, University of Medicine, Miami, FL 33136, USA 7 1st Medical Department, Johannes Gutenberg University Mainz, D-55131 Mainz, Germany 8 Institute of Clinical Neuroanatomy, Neuroscience Center, Goethe-University, Theodor-Stern-Kai 7, D-60590 Frankfurt, Germany 9 Neuropsychiatry and Laboratory of Molecular Psychiatry, Charite´ -Universita¨ tsmedizin Berlin, Schumannstr. 20-21, D-10117 Berlin, Germany 10 Institute of Genetics, Centre for Molecular Medicine (CMMC), and Cologne Excellence Cluster on Cellular Stress Responses in Aging-Associated Diseases (CECAD), University of Cologne, Zu¨ lpicher Str. -

Nerve Growth Factor: a Mitogenic Signal for Retinal Miiller Glial Cells
~,, ~ BRAIN ,, RESEARCH ELSEVIER Brain Research 64t~ {1994) 260-264 Research Report Nerve growth factor: a mitogenic signal for retinal Miiller glial cells Tsunehiko Ikeda, Donald G. Puro * Departments of Ophthalmolo~' and Physiology, Uni~'ersity of Michigan, W.K. Kellogg Eye Center, 1000 Wall Street, Ann Arbor, MI 48105, USA (Accepted 15 March 1994) Abstract Knowledge of the effects of nerve growth factor (NGF) on glia is limited. A CNS site where NGF-glial interactions may occur is the retina. NGF is endogenous to the retina, and the retinal Miiller glial cells have NGF receptors. Here, wc examined the possibility that NGF may be a mitogen for Miiller glial cells, which often proliferate in response to pathophysiological conditions. Experiments were performed on cultured glial cells from the adult human retina. Exposure of cultured Miiller glial cells to 2.5 S NGF under serum-free conditions resulted in a concentration-dependent increase in cell number and bromodeoxyuridinc incorporation into nuclei. The half-maximally effective concentration was 0.04 ng/ml (1.5 pM), consistent with activation of high affinity NGF receptors. K252a, a blocker of the neurotrophin family of tyrosine kinase-linked receptors, potently inhibited the proliferative effect of NGF. Transforming growth factor /3-2, another growth factor endogenous to the retina, inhibited the mitogenic response to NGF. These findings indicate that human Miiller glial cells in culture express functional NGF receptors and that the response of Mfiller cells to NGF can be modulated by other growth factors. Key words: Neurotrophin; Transforming growth factor/3-2; Gila; Proliferation; trk receptor 1. Introduction cellular event associated with irreversible loss of func- tion caused by detachment of the neural retina from It is established that certain neurons in the PNS and the underlying retinal pigment epithelium [5,7,29]. -

RT² Profiler PCR Array (Rotor-Gene® Format) Human Neurotrophins & Receptors
RT² Profiler PCR Array (Rotor-Gene® Format) Human Neurotrophins & Receptors Cat. no. 330231 PAHS-031ZR For pathway expression analysis Format For use with the following real-time cyclers RT² Profiler PCR Array, Rotor-Gene Q, other Rotor-Gene cyclers Format R Description The Human Neurotrophin & Receptors RT² Profiler PCR Array profiles the expression of 84 genes related to neuronal processes. Neurotrophic signaling molecules are represented on this array including neurotrophins and neuropeptides along with their receptors. Genes involved in the normal functions of the neuronal system including neuronal cell growth and differentiation and neuronal regeneration and survival are included. The cytokines and receptors involved in neuronal signaling are contained on this array along with genes involved in the transmission of nerve impulses, genes involved in neuronal apoptosis in response to neurotrophic factors and transcription factors and regulators indicative of the activation pathways downstream of the neuronal system. Using real-time PCR, you can easily and reliably analyze expression of a focused panel of genes related to the neuronal system with this array. For further details, consult the RT² Profiler PCR Array Handbook. Shipping and storage RT² Profiler PCR Arrays in the Rotor-Gene format are shipped at ambient temperature, on dry ice, or blue ice packs depending on destination and accompanying products. For long term storage, keep plates at –20°C. Note: Ensure that you have the correct RT² Profiler PCR Array format for your real-time cycler (see table above). Note: Open the package and store the products appropriately immediately on receipt. Sample & Assay Technologies Array layout The 96 real-time assays in the Rotor-Gene format are located in wells 1–96 of the Rotor-Disc™ (plate A1–A12=Rotor-Disc 1–12, plate B1–B12=Rotor-Disc 13–24, etc.).