Differential Metabolic Activity and Discovery of Therapeutic Targets Using Summarized Metabolic Pathway Models
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Enzyme DHRS7
Toward the identification of a function of the “orphan” enzyme DHRS7 Inauguraldissertation zur Erlangung der Würde eines Doktors der Philosophie vorgelegt der Philosophisch-Naturwissenschaftlichen Fakultät der Universität Basel von Selene Araya, aus Lugano, Tessin Basel, 2018 Originaldokument gespeichert auf dem Dokumentenserver der Universität Basel edoc.unibas.ch Genehmigt von der Philosophisch-Naturwissenschaftlichen Fakultät auf Antrag von Prof. Dr. Alex Odermatt (Fakultätsverantwortlicher) und Prof. Dr. Michael Arand (Korreferent) Basel, den 26.6.2018 ________________________ Dekan Prof. Dr. Martin Spiess I. List of Abbreviations 3α/βAdiol 3α/β-Androstanediol (5α-Androstane-3α/β,17β-diol) 3α/βHSD 3α/β-hydroxysteroid dehydrogenase 17β-HSD 17β-Hydroxysteroid Dehydrogenase 17αOHProg 17α-Hydroxyprogesterone 20α/βOHProg 20α/β-Hydroxyprogesterone 17α,20α/βdiOHProg 20α/βdihydroxyprogesterone ADT Androgen deprivation therapy ANOVA Analysis of variance AR Androgen Receptor AKR Aldo-Keto Reductase ATCC American Type Culture Collection CAM Cell Adhesion Molecule CYP Cytochrome P450 CBR1 Carbonyl reductase 1 CRPC Castration resistant prostate cancer Ct-value Cycle threshold-value DHRS7 (B/C) Dehydrogenase/Reductase Short Chain Dehydrogenase Family Member 7 (B/C) DHEA Dehydroepiandrosterone DHP Dehydroprogesterone DHT 5α-Dihydrotestosterone DMEM Dulbecco's Modified Eagle's Medium DMSO Dimethyl Sulfoxide DTT Dithiothreitol E1 Estrone E2 Estradiol ECM Extracellular Membrane EDTA Ethylenediaminetetraacetic acid EMT Epithelial-mesenchymal transition ER Endoplasmic Reticulum ERα/β Estrogen Receptor α/β FBS Fetal Bovine Serum 3 FDR False discovery rate FGF Fibroblast growth factor HEPES 4-(2-Hydroxyethyl)-1-Piperazineethanesulfonic Acid HMDB Human Metabolome Database HPLC High Performance Liquid Chromatography HSD Hydroxysteroid Dehydrogenase IC50 Half-Maximal Inhibitory Concentration LNCaP Lymph node carcinoma of the prostate mRNA Messenger Ribonucleic Acid n.d. -

Gene Networks Activated by Specific Patterns of Action Potentials in Dorsal Root Ganglia Neurons Received: 10 August 2016 Philip R
www.nature.com/scientificreports OPEN Gene networks activated by specific patterns of action potentials in dorsal root ganglia neurons Received: 10 August 2016 Philip R. Lee1,*, Jonathan E. Cohen1,*, Dumitru A. Iacobas2,3, Sanda Iacobas2 & Accepted: 23 January 2017 R. Douglas Fields1 Published: 03 March 2017 Gene regulatory networks underlie the long-term changes in cell specification, growth of synaptic connections, and adaptation that occur throughout neonatal and postnatal life. Here we show that the transcriptional response in neurons is exquisitely sensitive to the temporal nature of action potential firing patterns. Neurons were electrically stimulated with the same number of action potentials, but with different inter-burst intervals. We found that these subtle alterations in the timing of action potential firing differentially regulates hundreds of genes, across many functional categories, through the activation or repression of distinct transcriptional networks. Our results demonstrate that the transcriptional response in neurons to environmental stimuli, coded in the pattern of action potential firing, can be very sensitive to the temporal nature of action potential delivery rather than the intensity of stimulation or the total number of action potentials delivered. These data identify temporal kinetics of action potential firing as critical components regulating intracellular signalling pathways and gene expression in neurons to extracellular cues during early development and throughout life. Adaptation in the nervous system in response to external stimuli requires synthesis of new gene products in order to elicit long lasting changes in processes such as development, response to injury, learning, and memory1. Information in the environment is coded in the pattern of action-potential firing, therefore gene transcription must be regulated by the pattern of neuronal firing. -

Identification of Expression Qtls Targeting Candidate Genes For
ISSN: 2378-3648 Salleh et al. J Genet Genome Res 2018, 5:035 DOI: 10.23937/2378-3648/1410035 Volume 5 | Issue 1 Journal of Open Access Genetics and Genome Research RESEARCH ARTICLE Identification of Expression QTLs Targeting Candidate Genes for Residual Feed Intake in Dairy Cattle Using Systems Genomics Salleh MS1,2, Mazzoni G2, Nielsen MO1, Løvendahl P3 and Kadarmideen HN2,4* 1Department of Veterinary and Animal Sciences, Faculty of Health and Medical Sciences, University of Copenhagen, Denmark Check for 2Department of Bio and Health Informatics, Technical University of Denmark, Lyngby, Denmark updates 3Department of Molecular Biology and Genetics-Center for Quantitative Genetics and Genomics, Aarhus University, AU Foulum, Tjele, Denmark 4Department of Applied Mathematics and Computer Science, Technical University of Denmark, Lyngby, Denmark *Corresponding author: Kadarmideen HN, Department of Applied Mathematics and Computer Science, Technical University of Denmark, DK-2800, Kgs. Lyngby, Denmark, E-mail: [email protected] Abstract body weight gain and net merit). The eQTLs and biological pathways identified in this study improve our understanding Background: Residual feed intake (RFI) is the difference of the complex biological and genetic mechanisms that de- between actual and predicted feed intake and an important termine FE traits in dairy cattle. The identified eQTLs/genet- factor determining feed efficiency (FE). Recently, 170 can- ic variants can potentially be used in new genomic selection didate genes were associated with RFI, but no expression methods that include biological/functional information on quantitative trait loci (eQTL) mapping has hitherto been per- SNPs. formed on FE related genes in dairy cows. In this study, an integrative systems genetics approach was applied to map Keywords eQTLs in Holstein and Jersey cows fed two different diets to eQTL, RNA-seq, Genotype, Data integration, Systems improve identification of candidate genes for FE. -

Cystatin-B Negatively Regulates the Malignant Characteristics of Oral Squamous Cell Carcinoma Possibly Via the Epithelium Proliferation/ Differentiation Program
ORIGINAL RESEARCH published: 24 August 2021 doi: 10.3389/fonc.2021.707066 Cystatin-B Negatively Regulates the Malignant Characteristics of Oral Squamous Cell Carcinoma Possibly Via the Epithelium Proliferation/ Differentiation Program Tian-Tian Xu 1, Xiao-Wen Zeng 1, Xin-Hong Wang 2, Lu-Xi Yang 1, Gang Luo 1* and Ting Yu 1* 1 Department of Periodontics, Affiliated Stomatology Hospital of Guangzhou Medical University, Guangzhou Key Laboratory of Basic and Applied Research of Oral Regenerative Medicine, Guangzhou, China, 2 Department of Oral Pathology and Medicine, Affiliated Stomatology Hospital of Guangzhou Medical University, Guangzhou Key Laboratory of Basic and Applied Research of Oral Regenerative Medicine, Guangzhou, China Disturbance in the proteolytic process is one of the malignant signs of tumors. Proteolysis Edited by: Eva Csosz, is highly orchestrated by cysteine cathepsin and its inhibitors. Cystatin-B (CSTB) is a University of Debrecen, Hungary general cysteine cathepsin inhibitor that prevents cysteine cathepsin from leaking from Reviewed by: lysosomes and causing inappropriate proteolysis. Our study found that CSTB was Csongor Kiss, downregulated in both oral squamous cell carcinoma (OSCC) tissues and cells University of Debrecen, Hungary Gergely Nagy, compared with normal controls. Immunohistochemical analysis showed that CSTB was University of Debrecen, Hungary mainly distributed in the epithelial structure of OSCC tissues, and its expression intensity *Correspondence: was related to the grade classification. A correlation analysis between CSTB and clinical Gang Luo [email protected] prognosis was performed using gene expression data and clinical information acquired Ting Yu from The Cancer Genome Atlas (TCGA) database. Patients with lower expression levels of [email protected] CSTB had shorter disease-free survival times and poorer clinicopathological features Specialty section: (e.g., lymph node metastases, perineural invasion, low degree of differentiation, and This article was submitted to advanced tumor stage). -

Product Size GOT1 P00504 F CAAGCTGT
Table S1. List of primer sequences for RT-qPCR. Gene Product Uniprot ID F/R Sequence(5’-3’) name size GOT1 P00504 F CAAGCTGTCAAGCTGCTGTC 71 R CGTGGAGGAAAGCTAGCAAC OGDHL E1BTL0 F CCCTTCTCACTTGGAAGCAG 81 R CCTGCAGTATCCCCTCGATA UGT2A1 F1NMB3 F GGAGCAAAGCACTTGAGACC 93 R GGCTGCACAGATGAACAAGA GART P21872 F GGAGATGGCTCGGACATTTA 90 R TTCTGCACATCCTTGAGCAC GSTT1L E1BUB6 F GTGCTACCGAGGAGCTGAAC 105 R CTACGAGGTCTGCCAAGGAG IARS Q5ZKA2 F GACAGGTTTCCTGGCATTGT 148 R GGGCTTGATGAACAACACCT RARS Q5ZM11 F TCATTGCTCACCTGCAAGAC 146 R CAGCACCACACATTGGTAGG GSS F1NLE4 F ACTGGATGTGGGTGAAGAGG 89 R CTCCTTCTCGCTGTGGTTTC CYP2D6 F1NJG4 F AGGAGAAAGGAGGCAGAAGC 113 R TGTTGCTCCAAGATGACAGC GAPDH P00356 F GACGTGCAGCAGGAACACTA 112 R CTTGGACTTTGCCAGAGAGG Table S2. List of differentially expressed proteins during chronic heat stress. score name Description MW PI CC CH Down regulated by chronic heat stress A2M Uncharacterized protein 158 1 0.35 6.62 A2ML4 Uncharacterized protein 163 1 0.09 6.37 ABCA8 Uncharacterized protein 185 1 0.43 7.08 ABCB1 Uncharacterized protein 152 1 0.47 8.43 ACOX2 Cluster of Acyl-coenzyme A oxidase 75 1 0.21 8 ACTN1 Alpha-actinin-1 102 1 0.37 5.55 ALDOC Cluster of Fructose-bisphosphate aldolase 39 1 0.5 6.64 AMDHD1 Cluster of Uncharacterized protein 37 1 0.04 6.76 AMT Aminomethyltransferase, mitochondrial 42 1 0.29 9.14 AP1B1 AP complex subunit beta 103 1 0.15 5.16 APOA1BP NAD(P)H-hydrate epimerase 32 1 0.4 8.62 ARPC1A Actin-related protein 2/3 complex subunit 42 1 0.34 8.31 ASS1 Argininosuccinate synthase 47 1 0.04 6.67 ATP2A2 Cluster of Calcium-transporting -

Open Full Page
Published OnlineFirst November 2, 2012; DOI: 10.1158/2159-8290.CD-12-0031 RESEARCH ARTICLE Targeting C4-Demethylating Genes in the Cholesterol Pathway Sensitizes Cancer Cells to EGF Receptor Inhibitors via Increased EGF Receptor Degradation Anna Sukhanova 1 , Andrey Gorin 1 , Ilya G. Serebriiskii 1 , Linara Gabitova 1 , Hui Zheng 1 , Diana Restifo 1 , Brian L. Egleston 2 , David Cunningham 3 , Tetyana Bagnyukova 1 , Hanqing Liu 1 , Anna Nikonova 1 , Gregory P. Adams 1 , Yan Zhou 2 , Dong-Hua Yang 1 , Ranee Mehra 1 , Barbara Burtness 1 , Kathy Q. Cai 1 , Andres Klein-Szanto 1 , Lisa E. Kratz 4 , Richard I. Kelley 4 , Louis M. Weiner 5 , Gail E. Herman 3 , Erica A. Golemis 1 , and Igor Astsaturov 1 Downloaded from cancerdiscovery.aacrjournals.org on September 24, 2021. © 2013 American Association for Cancer Research. Published OnlineFirst November 2, 2012; DOI: 10.1158/2159-8290.CD-12-0031 ABSTRACT Persistent signaling by the oncogenic EGF receptor (EGFR) is a major source of cancer resistance to EGFR targeting. We established that inactivation of 2 sterol biosynthesis pathway genes, SC4MOL (sterol C4-methyl oxidase–like) and its partner, NSDHL (NADP-dependent steroid dehydrogenase–like), sensitized tumor cells to EGFR inhibitors. Bioinfor- matics modeling of interactions for the sterol pathway genes in eukaryotes allowed us to hypothesize and then extensively validate an unexpected role for SC4MOL and NSDHL in controlling the signaling, vesicular traffi cking, and degradation of EGFR and its dimerization partners, ERBB2 and ERBB3. Meta- bolic block upstream of SC4MOL with ketoconazole or CYP51A1 siRNA rescued cancer cell viability and EGFR degradation. -
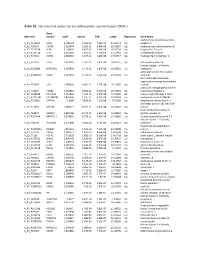
Table S1. the Statistical Metrics for Key Differentially Expressed Genes (Degs)
Table S1. The statistical metrics for key differentially expressed genes (DEGs) Gene Agilent Id Symbol logFC pValue FDR tvalue Regulation Gene Name oxidized low density lipoprotein A_24_P124624 OLR1 2.458429 1.19E-13 7.25E-10 24.04241 Up receptor 1 A_23_P90273 CHST8 2.622464 3.85E-12 6.96E-09 19.05867 Up carbohydrate sulfotransferase 8 A_23_P217528 KLF8 2.109007 4.85E-12 7.64E-09 18.76234 Up Kruppel like factor 8 A_23_P114740 CFH 2.651636 1.85E-11 1.79E-08 17.13652 Up complement factor H A_23_P34031 XAGE2 2.000935 2.04E-11 1.81E-08 17.02457 Up X antigen family member 2 A_23_P27332 TCF4 1.613097 2.32E-11 1.87E-08 16.87275 Up transcription factor 4 histone cluster 1 H1 family A_23_P250385 HIST1H1B 2.298658 2.47E-11 1.87E-08 16.80362 Up member b abnormal spindle microtubule A_33_P3288159 ASPM 2.162032 2.79E-11 2.01E-08 16.66292 Up assembly H19, imprinted maternally expressed transcript (non-protein A_24_P52697 H19 1.499364 4.09E-11 2.76E-08 16.23387 Up coding) potassium voltage-gated channel A_24_P31627 KCNB1 2.289689 6.65E-11 3.97E-08 15.70253 Up subfamily B member 1 A_23_P214168 COL12A1 2.155835 7.59E-11 4.15E-08 15.56005 Up collagen type XII alpha 1 chain A_33_P3271341 LOC388282 2.859496 7.61E-11 4.15E-08 15.55704 Up uncharacterized LOC388282 A_32_P150891 DIAPH3 2.2068 7.83E-11 4.22E-08 15.5268 Up diaphanous related formin 3 zinc finger protein 185 with LIM A_23_P11025 ZNF185 1.385721 8.74E-11 4.59E-08 15.41041 Up domain heat shock protein family B A_23_P96872 HSPB11 1.887166 8.94E-11 4.64E-08 15.38599 Up (small) member 11 A_23_P107454 -

Tissue-Based Alzheimer Gene Expression Markers–Comparison Of
Scheubert et al. BMC Bioinformatics 2012, 13:266 http://www.biomedcentral.com/1471-2105/13/266 RESEARCH ARTICLE Open Access Tissue-based Alzheimer gene expression markers–comparison of multiple machine learning approaches and investigation of redundancy in small biomarker sets Lena Scheubert1,3, Mitja Lustrekˇ 2, Rainer Schmidt3, Dirk Repsilber4* and Georg Fuellen3,5* Abstract Background: Alzheimer’s disease has been known for more than 100 years and the underlying molecular mechanisms are not yet completely understood. The identification of genes involved in the processes in Alzheimer affected brain is an important step towards such an understanding. Genes differentially expressed in diseased and healthy brains are promising candidates. Results: Based on microarray data we identify potential biomarkers as well as biomarker combinations using three feature selection methods: information gain, mean decrease accuracy of random forest and a wrapper of genetic algorithm and support vector machine (GA/SVM). Information gain and random forest are two commonly used methods. We compare their output to the results obtained from GA/SVM. GA/SVM is rarely used for the analysis of microarray data, but it is able to identify genes capable of classifying tissues into different classes at least as well as the two reference methods. Conclusion: Compared to the other methods, GA/SVM has the advantage of finding small, less redundant sets of genes that, in combination, show superior classification characteristics. The biological significance of the genes and gene pairs is discussed. Background could help both understanding the causes of the disease as Sporadic Alzheimer’s disease [1] is the most common well as suggest treatment options. -

Supplemental Information
1 1 Supplemental Information 2 3 Materials & Methods 4 Organisms. Anemones of the clonal Aiptasia strains CC7 (53) and H2 (54) were used 5 in this study, with H2 animals used only in the crosses to produce larvae for 6 transcriptome analyses (see below). Although CC7 and H2 have been described 7 previously as A. pallida and A. pulchella, respectively, recent global genotyping has 8 found two genetically distinct Aiptasia populations that do not conform to previous 9 species descriptions (5). Thus, we refer to both strains simply as Aiptasia in this paper. 10 Anemones were grown as described previously (19). Briefly, animals were raised in 11 a circulating artificial seawater (ASW) system at ~25ºC with 20-40 µmol photons m-2 s-1 12 of photosynthetically active radiation on a 12 h:12 h light:dark cycle and fed freshly 13 hatched Artemia (brine-shrimp) nauplii approximately twice per week. To generate 14 aposymbiotic anemones (i.e., animals without dinoflagellate symbionts), anemones were 15 placed in a polycarbonate tub and subjected to multiple repetitions of the following cycle: 16 cold-shocking by addition of 4ºC ASW and incubation at 4ºC for 4 h, followed by 1-2 d at 17 ~25ºC in ASW containing the photosynthesis inhibitor diuron (Sigma-Aldrich #D2425) at 18 50 µM (lighting approximately as above). The putatively aposymbiotic anemones were 19 then maintained for ≥1 month in ASW at ~25ºC in the light (as above) with feeding (as 20 above, with water changes on the days following feeding) to test for possible 21 repopulation of the animals by any residual dinoflagellates. -

Genetic Variation in Offspring Indirectly Influences the Quality of Maternal Behaviour in Mice David George Ashbrook*, Beatrice Gini, Reinmar Hager*
RESEARCH ARTICLE Genetic variation in offspring indirectly influences the quality of maternal behaviour in mice David George Ashbrook*, Beatrice Gini, Reinmar Hager* Computational and Evolutionary Biology, Faculty of Life Sciences, University of Manchester, Manchester, United Kingdom Abstract Conflict over parental investment between parent and offspring is predicted to lead to selection on genes expressed in offspring for traits influencing maternal investment, and on parentally expressed genes affecting offspring behaviour. However, the specific genetic variants that indirectly modify maternal or offspring behaviour remain largely unknown. Using a cross- fostered population of mice, we map maternal behaviour in genetically uniform mothers as a function of genetic variation in offspring and identify loci on offspring chromosomes 5 and 7 that modify maternal behaviour. Conversely, we found that genetic variation among mothers influences offspring development, independent of offspring genotype. Offspring solicitation and maternal behaviour show signs of coadaptation as they are negatively correlated between mothers and their biological offspring, which may be linked to costs of increased solicitation on growth found in our study. Overall, our results show levels of parental provisioning and offspring solicitation are unique to specific genotypes. DOI: 10.7554/eLife.11814.001 *For correspondence: david. [email protected]. ac.uk (DGA); Reinmar.Hager@ Introduction manchester.ac.uk (RH) The close interaction between mother and offspring in mammals is fundamental to offspring devel- Competing interests: The opment and fitness. However, parent and offspring are in conflict over how much parents should authors declare that no invest in their young where offspring typically demand more than is optimal for the parent (Triv- competing interests exist. -

Dual Regulatory Switch Through Interactions of Tcf7l2/Tcf4 with Stage-Specific Partners Propels Oligodendroglial Maturation
ARTICLE Received 19 Jun 2015 | Accepted 28 Jan 2016 | Published 9 Mar 2016 DOI: 10.1038/ncomms10883 OPEN Dual regulatory switch through interactions of Tcf7l2/Tcf4 with stage-specific partners propels oligodendroglial maturation Chuntao Zhao1,2,*, Yaqi Deng1,2,*, Lei Liu1,3,*, Kun Yu1, Liguo Zhang2, Haibo Wang2, Xuelian He2, Jincheng Wang2,4, Changqing Lu1, Laiman N. Wu2, Qinjie Weng4, Meng Mao3, Jianrong Li5, Johan H. van Es6, Mei Xin2, Lee Parry7, Steven A. Goldman8, Hans Clevers6 & Q. Richard Lu2,3,9 Constitutive activation of Wnt/b-catenin inhibits oligodendrocyte myelination. Tcf7l2/Tcf4, a b-catenin transcriptional partner, is required for oligodendrocyte differentiation. How Tcf7l2 modifies b-catenin signalling and controls myelination remains elusive. Here we define a stage-specific Tcf7l2-regulated transcriptional circuitry in initiating and sustaining oligodendrocyte differentiation. Multistage genome occupancy analyses reveal that Tcf7l2 serially cooperates with distinct co-regulators to control oligodendrocyte lineage progression. At the differentiation onset, Tcf7l2 interacts with a transcriptional co-repressor Kaiso/Zbtb33 to block b-catenin signalling. During oligodendrocyte maturation, Tcf7l2 recruits and cooperates with Sox10 to promote myelination. In that context, Tcf7l2 directly activates cholesterol biosynthesis genes and cholesterol supplementation partially rescues oligodendrocyte differentiation defects in Tcf712 mutants. Together, we identify stage-specific co-regulators Kaiso and Sox10 that sequentially interact with Tcf7l2 to coordinate the switch at the transitions of differentiation initiation and maturation during oligodendrocyte development, and point to a previously unrecognized role of Tcf7l2 in control of cholesterol biosynthesis for CNS myelinogenesis. 1 Department of Pediatrics, State Key Laboratory of Biotherapy, West China Second Hospital, Sichuan University, Collaborative Innovation Center forBiotherapy, Chengdu 610041, China. -

FMN Reduces Amyloid-β Toxicity in Yeast by Regulating Redox Status
ARTICLE https://doi.org/10.1038/s41467-020-14525-4 OPEN FMN reduces Amyloid-β toxicity in yeast by regulating redox status and cellular metabolism Xin Chen 1,2, Boyang Ji1,2, Xinxin Hao 3, Xiaowei Li1, Frederik Eisele 3, Thomas Nyström 3 & Dina Petranovic1,2* Alzheimer’s disease (AD) is defined by progressive neurodegeneration, with oligomerization and aggregation of amyloid-β peptides (Aβ) playing a pivotal role in its pathogenesis. In 1234567890():,; recent years, the yeast Saccharomyces cerevisiae has been successfully used to clarify the roles of different human proteins involved in neurodegeneration. Here, we report a genome-wide synthetic genetic interaction array to identify toxicity modifiers of Aβ42, using yeast as the model organism. We find that FMN1, the gene encoding riboflavin kinase, and its metabolic product flavin mononucleotide (FMN) reduce Aβ42 toxicity. Classic experimental analyses combined with RNAseq show the effects of FMN supplementation to include reducing misfolded protein load, altering cellular metabolism, increasing NADH/(NADH + NAD+) and NADPH/(NADPH + NADP+) ratios and increasing resistance to oxidative stress. Addition- ally, FMN supplementation modifies Htt103QP toxicity and α-synuclein toxicity in the humanized yeast. Our findings offer insights for reducing cytotoxicity of Aβ42, and potentially other misfolded proteins, via FMN-dependent cellular pathways. 1 Division of Systems and Synthetic Biology, Department of Biology and Biological Engineering, Chalmers University of Technology, SE41296 Gothenburg, Sweden.