Cryptic 7Q21 and 9P23 Deletions in a Patient with Apparently
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Analyses of Allele-Specific Gene Expression in Highly Divergent
ARTICLES Analyses of allele-specific gene expression in highly divergent mouse crosses identifies pervasive allelic imbalance James J Crowley1,10, Vasyl Zhabotynsky1,10, Wei Sun1,2,10, Shunping Huang3, Isa Kemal Pakatci3, Yunjung Kim1, Jeremy R Wang3, Andrew P Morgan1,4,5, John D Calaway1,4,5, David L Aylor1,9, Zaining Yun1, Timothy A Bell1,4,5, Ryan J Buus1,4,5, Mark E Calaway1,4,5, John P Didion1,4,5, Terry J Gooch1,4,5, Stephanie D Hansen1,4,5, Nashiya N Robinson1,4,5, Ginger D Shaw1,4,5, Jason S Spence1, Corey R Quackenbush1, Cordelia J Barrick1, Randal J Nonneman1, Kyungsu Kim2, James Xenakis2, Yuying Xie1, William Valdar1,4, Alan B Lenarcic1, Wei Wang3,9, Catherine E Welsh3, Chen-Ping Fu3, Zhaojun Zhang3, James Holt3, Zhishan Guo3, David W Threadgill6, Lisa M Tarantino7, Darla R Miller1,4,5, Fei Zou2,11, Leonard McMillan3,11, Patrick F Sullivan1,5,7,8,11 & Fernando Pardo-Manuel de Villena1,4,5,11 Complex human traits are influenced by variation in regulatory DNA through mechanisms that are not fully understood. Because regulatory elements are conserved between humans and mice, a thorough annotation of cis regulatory variants in mice could aid in further characterizing these mechanisms. Here we provide a detailed portrait of mouse gene expression across multiple tissues in a three-way diallel. Greater than 80% of mouse genes have cis regulatory variation. Effects from these variants influence complex traits and usually extend to the human ortholog. Further, we estimate that at least one in every thousand SNPs creates a cis regulatory effect. -

Murine MPDZ-Linked Hydrocephalus Is Caused by Hyperpermeability of the Choroid Plexus
Thomas Jefferson University Jefferson Digital Commons Cardeza Foundation for Hematologic Research Sidney Kimmel Medical College 1-1-2019 Murine MPDZ-linked hydrocephalus is caused by hyperpermeability of the choroid plexus. Junning Yang Thomas Jefferson University Claire Simonneau Thomas Jefferson University Robert Kilker Thomas Jefferson University Laura Oakley ThomasFollow this Jeff anderson additional University works at: https://jdc.jefferson.edu/cardeza_foundation Part of the Medical Pathology Commons Matthew D. Byrne ThomasLet us Jeff knowerson Univ howersity access to this document benefits ouy Recommended Citation See next page for additional authors Yang, Junning; Simonneau, Claire; Kilker, Robert; Oakley, Laura; Byrne, Matthew D.; Nichtova, Zuzana; Stefanescu, Ioana; Pardeep-Kumar, Fnu; Tripathi, Sushil; Londin, Eric; Saugier-Veber, Pascale; Willard, Belinda; Thakur, Mathew; Pickup, Stephen; Ishikawa, Hiroshi; Schroten, Horst; Smeyne, Richard; and Horowitz, Arie, "Murine MPDZ-linked hydrocephalus is caused by hyperpermeability of the choroid plexus." (2019). Cardeza Foundation for Hematologic Research. Paper 49. https://jdc.jefferson.edu/cardeza_foundation/49 This Article is brought to you for free and open access by the Jefferson Digital Commons. The Jefferson Digital Commons is a service of Thomas Jefferson University's Center for Teaching and Learning (CTL). The Commons is a showcase for Jefferson books and journals, peer-reviewed scholarly publications, unique historical collections from the University archives, and teaching tools. The Jefferson Digital Commons allows researchers and interested readers anywhere in the world to learn about and keep up to date with Jefferson scholarship. This article has been accepted for inclusion in Cardeza Foundation for Hematologic Research by an authorized administrator of the Jefferson Digital Commons. For more information, please contact: [email protected]. -

Gene Section Short Communication
Atlas of Genetics and Cytogenetics in Oncology and Haematology OPEN ACCESS JOURNAL INIST-CNRS Gene Section Short Communication TYRP1 (tyrosinase-related protein 1) Kunal Ray, Mainak Sengupta, Sampurna Ghosh Academy of Scientific and Innovative Research (AcSIR), Campus at CSIR - Central Road Research Institute, Mathura Road, New Delhi - 110 025, [email protected] (KR); University of Calcutta, Department of Genetics, 35, Ballygunge Circular Road, Kolkata - 700 019, [email protected]); [email protected] (MS, SG) India. Published in Atlas Database: April 2016 Online updated version : http://AtlasGeneticsOncology.org/Genes/TYRP1ID46370ch9p23.html Printable original version : http://documents.irevues.inist.fr/bitstream/handle/2042/68125/04-2016-TYRP1ID46370ch9p23.pdf DOI: 10.4267/2042/68125 This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 2.0 France Licence. © 2016 Atlas of Genetics and Cytogenetics in Oncology and Haematology Abstract Location: 9p23 TYRP1 gene, having a chromosomal location of 9p23, encodes a melanosomal enzyme belonging to DNA/RNA the tyrosinase family. TYRP1 catalyses oxidation of 5,6-dihydroxyindole-2-carboxylic acid (DHICA) Description into indole-5,6-quinone-2-carboxylic acid. TYRP1 In Chromosome 9, the 24,852 bases long gene starts is also thought to play a role in stabilizing tyrosinase from12,685,439 bp from pter and ends at 12,710,290 and modulates its catalytic activity, in maintenance bp from pter; Orientation: Plus strand. The gene of melanosome structure, affecting melanocyte contains 8 exons and spans ~24.8 kb of the genome. proliferation and melanocyte cell death. Defects in this gene cause oculocutaneous albinism type III; Transcription OCA III (also known as rufous oculocutaneous The gene encodes a 2876 bp mRNA. -

Dog Coat Colour Genetics: a Review Date Published Online: 31/08/2020; 1,2 1 1 3 Rashid Saif *, Ali Iftekhar , Fatima Asif , Mohammad Suliman Alghanem
www.als-journal.com/ ISSN 2310-5380/ August 2020 Review Article Advancements in Life Sciences – International Quarterly Journal of Biological Sciences ARTICLE INFO Open Access Date Received: 02/05/2020; Date Revised: 20/08/2020; Dog Coat Colour Genetics: A Review Date Published Online: 31/08/2020; 1,2 1 1 3 Rashid Saif *, Ali Iftekhar , Fatima Asif , Mohammad Suliman Alghanem Authors’ Affiliation: 1. Institute of Abstract Biotechnology, Gulab Devi Educational anis lupus familiaris is one of the most beloved pet species with hundreds of world-wide recognized Complex, Lahore - Pakistan breeds, which can be differentiated from each other by specific morphological, behavioral and adoptive 2. Decode Genomics, traits. Morphological characteristics of dog breeds get more attention which can be defined mostly by 323-D, Town II, coat color and its texture, and considered to be incredibly lucrative traits in this valued species. Although Punjab University C Employees Housing the genetic foundation of coat color has been well stated in the literature, but still very little is known about the Scheme, Lahore - growth pattern, hair length and curly coat trait genes. Skin pigmentation is determined by eumelanin and Pakistan 3. Department of pheomelanin switching phenomenon which is under the control of Melanocortin 1 Receptor and Agouti Signaling Biology, Tabuk Protein genes. Genetic variations in the genes involved in pigmentation pathway provide basic understanding of University - Kingdom melanocortin physiology and evolutionary adaptation of this trait. So in this review, we highlighted, gathered and of Saudi Arabia comprehend the genetic mutations, associated and likely to be associated variants in the genes involved in the coat color and texture trait along with their phenotypes. -
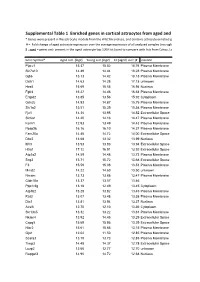
Supplemental Table 1 Enriched Genes in Cortical Astrocytes from Aged
Supplemental Table 1 Enriched genes in cortical astrocytes from aged and young-adult mice * Genes were present in the astrocyte module from the WGCNA analysis, and contains astrocyte enriched genes compared to microglia and oligodendrocytes # = Fold change of aged astrocyte expression over the average expression of all analyzed samples (microglia, astrocytes: young, old, with and without myelin contamination) $ ; aged = genes only present in the aged astrocyte top 1000 list (used to compare with lists from Cahoy, Lovatt, Doyle; see Fig. 4B), all = genes present in all astrocyte top 1000 lists Gene Symbol* Aged astr. (log2) Young astr.(log2) FC (aged/ aver.)# Location Ptprz1 15.37 15.02 18.76 Plasma Membrane Slc7a10 14.49 14.44 18.28 Plasma Membrane Gjb6 15.13 14.42 18.18 Plasma Membrane Dclk1 14.63 14.28 17.18 unknown Hes5 15.69 15.55 16.94 Nucleus Fgfr3 15.27 14.46 16.54 Plasma Membrane Entpd2 13.85 13.56 15.92 Cytoplasm Grin2c 14.93 14.87 15.75 Plasma Membrane Slc1a2 15.51 15.39 15.58 Plasma Membrane Fjx1 14.36 13.98 14.52 Extracellular Space Slc6a1 14.20 14.16 14.47 Plasma Membrane Kcnk1 12.93 13.49 14.43 Plasma Membrane Ppap2b 16.16 16.10 14.37 Plasma Membrane Fam20a 14.48 14.72 14.00 Extracellular Space Dbx2 13.68 13.32 13.99 Nucleus Itih3 13.93 13.93 13.94 Extracellular Space Htra1 17.12 16.91 13.92 Extracellular Space Atp1a2 14.59 14.48 13.73 Plasma Membrane Scg3 15.71 15.72 13.68 Extracellular Space F3 15.59 15.08 13.51 Plasma Membrane Mmd2 14.22 14.60 13.50 unknown Nrcam 13.73 13.88 13.47 Plasma Membrane Cldn10a 13.37 13.57 13.46 -

The Utility of Genetic Risk Scores in Predicting the Onset of Stroke March 2021 6
DOT/FAA/AM-21/24 Office of Aerospace Medicine Washington, DC 20591 The Utility of Genetic Risk Scores in Predicting the Onset of Stroke Diana Judith Monroy Rios, M.D1 and Scott J. Nicholson, Ph.D.2 1. KR 30 # 45-03 University Campus, Building 471, 5th Floor, Office 510 Bogotá D.C. Colombia 2. FAA Civil Aerospace Medical Institute, 6500 S. MacArthur Blvd Rm. 354, Oklahoma City, OK 73125 March 2021 NOTICE This document is disseminated under the sponsorship of the U.S. Department of Transportation in the interest of information exchange. The United States Government assumes no liability for the contents thereof. _________________ This publication and all Office of Aerospace Medicine technical reports are available in full-text from the Civil Aerospace Medical Institute’s publications Web site: (www.faa.gov/go/oamtechreports) Technical Report Documentation Page 1. Report No. 2. Government Accession No. 3. Recipient's Catalog No. DOT/FAA/AM-21/24 4. Title and Subtitle 5. Report Date March 2021 The Utility of Genetic Risk Scores in Predicting the Onset of Stroke 6. Performing Organization Code 7. Author(s) 8. Performing Organization Report No. Diana Judith Monroy Rios M.D1, and Scott J. Nicholson, Ph.D.2 9. Performing Organization Name and Address 10. Work Unit No. (TRAIS) 1 KR 30 # 45-03 University Campus, Building 471, 5th Floor, Office 510, Bogotá D.C. Colombia 11. Contract or Grant No. 2 FAA Civil Aerospace Medical Institute, 6500 S. MacArthur Blvd Rm. 354, Oklahoma City, OK 73125 12. Sponsoring Agency name and Address 13. Type of Report and Period Covered Office of Aerospace Medicine Federal Aviation Administration 800 Independence Ave., S.W. -

New Approaches to Functional Process Discovery in HPV 16-Associated Cervical Cancer Cells by Gene Ontology
Cancer Research and Treatment 2003;35(4):304-313 New Approaches to Functional Process Discovery in HPV 16-Associated Cervical Cancer Cells by Gene Ontology Yong-Wan Kim, Ph.D.1, Min-Je Suh, M.S.1, Jin-Sik Bae, M.S.1, Su Mi Bae, M.S.1, Joo Hee Yoon, M.D.2, Soo Young Hur, M.D.2, Jae Hoon Kim, M.D.2, Duck Young Ro, M.D.2, Joon Mo Lee, M.D.2, Sung Eun Namkoong, M.D.2, Chong Kook Kim, Ph.D.3 and Woong Shick Ahn, M.D.2 1Catholic Research Institutes of Medical Science, 2Department of Obstetrics and Gynecology, College of Medicine, The Catholic University of Korea, Seoul; 3College of Pharmacy, Seoul National University, Seoul, Korea Purpose: This study utilized both mRNA differential significant genes of unknown function affected by the display and the Gene Ontology (GO) analysis to char- HPV-16-derived pathway. The GO analysis suggested that acterize the multiple interactions of a number of genes the cervical cancer cells underwent repression of the with gene expression profiles involved in the HPV-16- cancer-specific cell adhesive properties. Also, genes induced cervical carcinogenesis. belonging to DNA metabolism, such as DNA repair and Materials and Methods: mRNA differential displays, replication, were strongly down-regulated, whereas sig- with HPV-16 positive cervical cancer cell line (SiHa), and nificant increases were shown in the protein degradation normal human keratinocyte cell line (HaCaT) as a con- and synthesis. trol, were used. Each human gene has several biological Conclusion: The GO analysis can overcome the com- functions in the Gene Ontology; therefore, several func- plexity of the gene expression profile of the HPV-16- tions of each gene were chosen to establish a powerful associated pathway, identify several cancer-specific cel- cervical carcinogenesis pathway. -

Supplementary Table 3 Complete List of RNA-Sequencing Analysis of Gene Expression Changed by ≥ Tenfold Between Xenograft and Cells Cultured in 10%O2
Supplementary Table 3 Complete list of RNA-Sequencing analysis of gene expression changed by ≥ tenfold between xenograft and cells cultured in 10%O2 Expr Log2 Ratio Symbol Entrez Gene Name (culture/xenograft) -7.182 PGM5 phosphoglucomutase 5 -6.883 GPBAR1 G protein-coupled bile acid receptor 1 -6.683 CPVL carboxypeptidase, vitellogenic like -6.398 MTMR9LP myotubularin related protein 9-like, pseudogene -6.131 SCN7A sodium voltage-gated channel alpha subunit 7 -6.115 POPDC2 popeye domain containing 2 -6.014 LGI1 leucine rich glioma inactivated 1 -5.86 SCN1A sodium voltage-gated channel alpha subunit 1 -5.713 C6 complement C6 -5.365 ANGPTL1 angiopoietin like 1 -5.327 TNN tenascin N -5.228 DHRS2 dehydrogenase/reductase 2 leucine rich repeat and fibronectin type III domain -5.115 LRFN2 containing 2 -5.076 FOXO6 forkhead box O6 -5.035 ETNPPL ethanolamine-phosphate phospho-lyase -4.993 MYO15A myosin XVA -4.972 IGF1 insulin like growth factor 1 -4.956 DLG2 discs large MAGUK scaffold protein 2 -4.86 SCML4 sex comb on midleg like 4 (Drosophila) Src homology 2 domain containing transforming -4.816 SHD protein D -4.764 PLP1 proteolipid protein 1 -4.764 TSPAN32 tetraspanin 32 -4.713 N4BP3 NEDD4 binding protein 3 -4.705 MYOC myocilin -4.646 CLEC3B C-type lectin domain family 3 member B -4.646 C7 complement C7 -4.62 TGM2 transglutaminase 2 -4.562 COL9A1 collagen type IX alpha 1 chain -4.55 SOSTDC1 sclerostin domain containing 1 -4.55 OGN osteoglycin -4.505 DAPL1 death associated protein like 1 -4.491 C10orf105 chromosome 10 open reading frame 105 -4.491 -

Multiple Cellular Proteins Interact with LEDGF/P75 Through a Conserved Unstructured Consensus Motif
ARTICLE Received 19 Jan 2015 | Accepted 1 Jul 2015 | Published 6 Aug 2015 DOI: 10.1038/ncomms8968 Multiple cellular proteins interact with LEDGF/p75 through a conserved unstructured consensus motif Petr Tesina1,2,3,*, Katerˇina Cˇerma´kova´4,*, Magdalena Horˇejsˇ´ı3, Katerˇina Procha´zkova´1, Milan Fa´bry3, Subhalakshmi Sharma4, Frauke Christ4, Jonas Demeulemeester4, Zeger Debyser4, Jan De Rijck4,**, Va´clav Veverka1,** & Pavlı´na Rˇeza´cˇova´1,3,** Lens epithelium-derived growth factor (LEDGF/p75) is an epigenetic reader and attractive therapeutic target involved in HIV integration and the development of mixed lineage leukaemia (MLL1) fusion-driven leukaemia. Besides HIV integrase and the MLL1-menin complex, LEDGF/p75 interacts with various cellular proteins via its integrase binding domain (IBD). Here we present structural characterization of IBD interactions with transcriptional repressor JPO2 and domesticated transposase PogZ, and show that the PogZ interaction is nearly identical to the interaction of LEDGF/p75 with MLL1. The interaction with the IBD is maintained by an intrinsically disordered IBD-binding motif (IBM) common to all known cellular partners of LEDGF/p75. In addition, based on IBM conservation, we identify and validate IWS1 as a novel LEDGF/p75 interaction partner. Our results also reveal how HIV integrase efficiently displaces cellular binding partners from LEDGF/p75. Finally, the similar binding modes of LEDGF/p75 interaction partners represent a new challenge for the development of selective interaction inhibitors. 1 Institute of Organic Chemistry and Biochemistry of the ASCR, v.v.i., Flemingovo nam. 2, 166 10 Prague, Czech Republic. 2 Department of Genetics and Microbiology, Faculty of Science, Charles University in Prague, Vinicna 5, 128 44 Prague, Czech Republic. -

Supplementary Table 1: Adhesion Genes Data Set
Supplementary Table 1: Adhesion genes data set PROBE Entrez Gene ID Celera Gene ID Gene_Symbol Gene_Name 160832 1 hCG201364.3 A1BG alpha-1-B glycoprotein 223658 1 hCG201364.3 A1BG alpha-1-B glycoprotein 212988 102 hCG40040.3 ADAM10 ADAM metallopeptidase domain 10 133411 4185 hCG28232.2 ADAM11 ADAM metallopeptidase domain 11 110695 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 195222 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 165344 8751 hCG20021.3 ADAM15 ADAM metallopeptidase domain 15 (metargidin) 189065 6868 null ADAM17 ADAM metallopeptidase domain 17 (tumor necrosis factor, alpha, converting enzyme) 108119 8728 hCG15398.4 ADAM19 ADAM metallopeptidase domain 19 (meltrin beta) 117763 8748 hCG20675.3 ADAM20 ADAM metallopeptidase domain 20 126448 8747 hCG1785634.2 ADAM21 ADAM metallopeptidase domain 21 208981 8747 hCG1785634.2|hCG2042897 ADAM21 ADAM metallopeptidase domain 21 180903 53616 hCG17212.4 ADAM22 ADAM metallopeptidase domain 22 177272 8745 hCG1811623.1 ADAM23 ADAM metallopeptidase domain 23 102384 10863 hCG1818505.1 ADAM28 ADAM metallopeptidase domain 28 119968 11086 hCG1786734.2 ADAM29 ADAM metallopeptidase domain 29 205542 11085 hCG1997196.1 ADAM30 ADAM metallopeptidase domain 30 148417 80332 hCG39255.4 ADAM33 ADAM metallopeptidase domain 33 140492 8756 hCG1789002.2 ADAM7 ADAM metallopeptidase domain 7 122603 101 hCG1816947.1 ADAM8 ADAM metallopeptidase domain 8 183965 8754 hCG1996391 ADAM9 ADAM metallopeptidase domain 9 (meltrin gamma) 129974 27299 hCG15447.3 ADAMDEC1 ADAM-like, -

Comparative Genome Mapping in the Sequence-Based Era: Early Experience with Human Chromosome 7
Downloaded from genome.cshlp.org on May 28, 2019 - Published by Cold Spring Harbor Laboratory Press First Glimpses/Report Comparative Genome Mapping in the Sequence-based Era: Early Experience with Human Chromosome 7 James W. Thomas,1 Tyrone J. Summers,1 Shih-Queen Lee-Lin,1 Valerie V. Braden Maduro,1 Jacquelyn R. Idol,1 Stephen D. Mastrian,1 Joseph F. Ryan,1 D. Curtis Jamison,1 and Eric D. Green1,2 1Genome Technology Branch, National Human Genome Research Institute, National Institutes of Health, Bethesda, Maryland 20892 USA The success of the ongoing Human Genome Project has resulted in accelerated plans for completing the human genome sequence and the earlier-than-anticipated initiation of efforts to sequence the mouse genome. As a complement to these efforts, we are utilizing the available human sequence to refine human-mouse comparative maps and to assemble sequence-ready mouse physical maps. Here we describe how the first glimpses of genomic sequence from human chromosome 7 are directly facilitating these activities. Specifically, we are actively enhancing the available human-mouse comparative map by analyzing human chromosome 7 sequence for the presence of orthologs of mapped mouse genes. Such orthologs can then be precisely positioned relative to mapped human STSs and other genes. The chromosome 7 sequence generated to date has allowed us to more than double the number of genes that can be placed on the comparative map. The latter effort reveals that human chromosome 7 is represented by at least 20 orthologous segments of DNA in the mouse genome. A second component of our program involves systematically analyzing the evolving human chromosome 7 sequence for the presence of matching mouse genes and expressed-sequence tags (ESTs). -

Comparative Analysis of Human Chromosome 7Q21 and Mouse
Downloaded from genome.cshlp.org on October 2, 2021 - Published by Cold Spring Harbor Laboratory Press Letter Comparative analysis of human chromosome 7q21 and mouse proximal chromosome 6 reveals a placental-specific imprinted gene, TFPI2/Tfpi2, which requires EHMT2 and EED for allelic-silencing David Monk,1,6 Alexandre Wagschal,2 Philippe Arnaud,2 Pari-Sima Mu¨ller,3 Layla Parker-Katiraee,4 Déborah Bourc’his,5 Stephen W. Scherer,4 Robert Feil,2 Philip Stanier,1 and Gudrun E. Moore1 1Institute of Child Health, London WC1N 1EH, United Kingdom; 2Institute of Molecular Genetics, CNRS UMR-5535 and University of Montpellier-II, 34293 Montpellier, France; 3Sir William Dunn School of Pathology, University of Oxford, Oxford OX1 3RE, United Kingdom; 4Center for Applied Genomics, The Hospital for Sick Children, Toronto M5G 1L7, Canada; 5Inserm U741, F-75251 Paris Cedex 05, France Genomic imprinting is a developmentally important mechanism that involves both differential DNA methylation and allelic histone modifications. Through detailed comparative characterization, a large imprinted domain mapping to chromosome 7q21 in humans and proximal chromosome 6 in mice was redefined. This domain is organized around a maternally methylated CpG island comprising the promoters of the adjacent PEG10 and SGCE imprinted genes. Examination of Dnmt3l−/+ conceptuses shows that imprinted expression for all genes of the cluster depends upon the germline methylation at this putative “imprinting control region” (ICR). Similarly as for other ICRs, we find its DNA-methylated allele to be associated with trimethylation of lysine 9 on histone H3 (H3K9me3) and trimethylation of lysine 20 on histone H4 (H4K20me3), whereas the transcriptionally active paternal allele is enriched in H3K4me2 and H3K9 acetylation.