Introduction to Proteins
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Heterogeneous Impacts of Protein-Stabilizing Osmolytes On
bioRxiv preprint doi: https://doi.org/10.1101/328922; this version posted May 23, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Heterogeneous Impacts of Protein-Stabilizing Osmolytes on Hydrophobic Interaction Mrinmoy Mukherjee and Jagannath Mondal⇤ Tata Institute of Fundamental Research Hyderabad, 500107 India E-mail: [email protected],+914020203091 Abstract Osmolytes’ mechanism of protecting proteins against denaturation is a longstanding puzzle, further complicated by the complex diversities inherent in protein sequences. An emergent approach in understanding osmolytes’ mechanism of action towards biopoly- mer has been to investigate osmolytes’ interplay with hydrophobic interaction, the ma- jor driving force of protein folding. However, the crucial question is whether all these protein-stabilizing osmolytes display a single unified mechanism towards hydrophobic interactions. By simulating the hydrophobic collapse of a macromolecule in aqueous solutions of two such osmoprotectants, Glycine and Trimethyl N-oxide (TMAO), both of which are known to stabilize protein’s folded conformation, we here demonstrate that these two osmolytes can impart mutually contrasting effects towards hydropho- bic interaction. While TMAO preserves its protectant nature across diverse range of polymer-osmolyte interactions, glycine is found to display an interesting cross-over from being a protectant at weaker polymer-osmolyte interaction to a denaturant of hydrophobicity at stronger polymer-osmolyte interactions. A preferential-interaction analysis reveals that a subtle balance of conformation-dependent exclusion/binding of ⇤To whom correspondence should be addressed 1 bioRxiv preprint doi: https://doi.org/10.1101/328922; this version posted May 23, 2018. -

Protein Structure & Folding
6 Protein Structure & Folding To understand protein folding In the last chapter we learned that proteins are composed of amino acids Goal as a chemical equilibrium. linked together by peptide bonds. We also learned that the twenty amino acids display a wide range of chemical properties. In this chapter we will see Objectives that how a protein folds is determined by its amino acid sequence and that the three-dimensional shape of a folded protein determines its function by After this chapter, you should be able to: the way it positions these amino acids. Finally, we will see that proteins fold • describe the four levels of protein because doing so minimizes Gibbs free energy and that this minimization structure and the thermodynamic involves both making the most favorable bonds and maximizing disorder. forces that stabilize them. • explain how entropy (S) and enthalpy Proteins exhibit four levels of structure (H) contribute to Gibbs free energy. • use the equation ΔG = ΔH – TΔS The structure of proteins can be broken down into four levels of to determine the dependence of organization. The first is primary structure, the linear sequence of amino the favorability of a reaction on acids in the polypeptide chain. By convention, the primary sequence is temperature. written in order from the amino acid at the N-terminus (by convention • explain the hydrophobic effect and its usually on the left) to the amino acid at the C-terminus. The second level role in protein folding. of protein structure, secondary structure, is the local conformation adopted by stretches of contiguous amino acids. -

A Network of Molecular Switches Controls the Activation of the Two-Component Response Regulator Ntrc
ARTICLE Received 8 Aug 2014 | Accepted 26 Apr 2015 | Published 15 Jun 2015 DOI: 10.1038/ncomms8283 A network of molecular switches controls the activation of the two-component response regulator NtrC Dan K. Vanatta1, Diwakar Shukla1,2,3, Morgan Lawrenz1 & Vijay S. Pande1,2 Recent successes in simulating protein structure and folding dynamics have demonstrated the power of molecular dynamics to predict the long timescale behaviour of proteins. Here, we extend and improve these methods to predict molecular switches that characterize conformational change pathways between the active and inactive state of nitrogen regulatory protein C (NtrC). By employing unbiased Markov state model-based molecular dynamics simulations, we construct a dynamic picture of the activation pathways of this key bacterial signalling protein that is consistent with experimental observations and predicts new mutants that could be used for validation of the mechanism. Moreover, these results suggest a novel mechanistic paradigm for conformational switching. 1 Department of Chemistry, Stanford University, Stanford, California 94305, USA. 2 SIMBIOS NIH Center for Biomedical Computation, Stanford University, Stanford, California 94305,USA. 3 Department of Chemical and Bimolecular Engineering, University of Illinois at Urbana-Champaign, Urbana, Illionios 61801, USA. Correspondence and requests for materials should be addressed to V.S.P. (email: [email protected]). NATURE COMMUNICATIONS | 6:7283 | DOI: 10.1038/ncomms8283 | www.nature.com/naturecommunications 1 & 2015 Macmillan Publishers Limited. All rights reserved. ARTICLE NATURE COMMUNICATIONS | DOI: 10.1038/ncomms8283 roteins involved in cellular signalling change their con- Results formation in response to changes in environment (input Simulations reveal molecular switches for NtrC activation. Psignal), such as ligand binding or chemical modification, MSMs for NtrC were built using snapshots along the simulated to control downstream cellular processes (output signal)1. -

Spring 2013 Lecture 23
CHM333 LECTURES 23: 3/25/13 SPRING 2013 Professor Christine Hrycyna LIPIDS III EFFECT OF CHOLESTEROL ON MEMBRANES: - Bulky rigid molecule - Moderates fluidity of membranes – both increases and decreases o Cholesterol in membranes DECREASES fluidity because it is rigid o Prevents crystallization (making solid) of fatty acyl side chains by fitting between them. Disrupts close packing of fatty acyl chains. Therefore, INCREASED fluidity BIOLOGICAL MEMBRANES CONTAIN PROTEINS AS WELL AS LIPIDS: - Proteins are 20-80% of cell membrane - Rest is lipid or carbohydrate; supramolecular assembly of lipid, protein and carbohydrate - Proteins are also distributed asymmetrically - TWO classes of Membrane Proteins: o Integral Membrane Proteins o Peripheral Membrane Proteins 178 CHM333 LECTURES 23: 3/25/13 SPRING 2013 Professor Christine Hrycyna - INTEGRAL MEMBRANE PROTEINS o Located WITHIN the lipid bilayer o Usually span the bilayer one or more times – called transmembrane (TM) proteins o Hydrophobic amino acids interact with fatty acid chains in the hydrophobic core of the membrane o Can be removed from the membrane with detergents like SDS – need to disrupt the hydrophobic interactions § Membrane Disruption Animation: o http://www.youtube.com/watch?v=AHT37pvcjc0 o Function: § Transporters – moving molecules into or out of cells or cell membranes § Receptors – transmitting signals from outside of the cell to the inside - β Barrel Integral Membrane Proteins § Barrel-shaped membrane protein that is made up of antiparallel β-strands with hydrophilic (interior) and hydrophobic (facing lipid tails). § So far found only in outer membranes of Gram-negative bacteria, cell wall of Gram-positive bacteria, and outer membranes of mitochondria and chloroplasts. 179 CHM333 LECTURES 23: 3/25/13 SPRING 2013 Professor Christine Hrycyna - α-Helical Membrane Proteins - Can cross the membrane once or many times and have multiple transmembrane segments. -
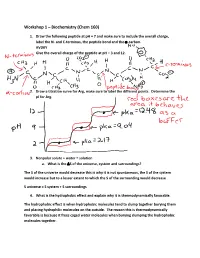
Workshop 1 – Biochemistry (Chem 160)
Workshop 1 – Biochemistry (Chem 160) 1. Draw the following peptide at pH = 7 and make sure to include the overall charge, label the N- and C-terminus, the peptide bond and the -carbon. AVDKY Give the overall charge of the peptide at pH = 3 and 12. 2. Draw a titration curve for Arg, make sure to label the different points. Determine the pI for Arg. 3. Nonpolar solute + water = solution a. What is the S of the universe, system and surroundings? The S of the universe would decrease this is why it is not spontaneous, the S of the system would increase but to a lesser extent to which the S of the surrounding would decrease S universe = S system + S surroundings 4. What is the hydrophobic effect and explain why it is thermodynamically favorable. The hydrophobic effect is when hydrophobic molecules tend to clump together burying them and placing hydrophilic molecules on the outside. The reason this is thermodynamically favorable is because it frees caged water molecules when burying clumping the hydrophobic molecules together. 5. Urea dissolves very readily in water, but the solution becomes very cold as the urea dissolves. How is this possible? Urea dissolves in water because when dissolving there is a net increase in entropy of the universe. The heat exchange, getting colder only reflects the enthalpy (H) component of the total energy change. The entropy change is high enough to offset the enthalpy component and to add up to an overall -G 6. A mutation that changes an alanine residue in the interior of a protein to valine is found to lead to a loss of activity. -

Site-Specific Tryptophan Labels Reveal Local Microsecond
molecules Article Site-Specific Tryptophan Labels Reveal Local Microsecond–Millisecond Motions of Dihydrofolate Reductase Morgan B. Vaughn 1,*, Chloe Biren 2, Qun Li 2, Ashwin Ragupathi 2 and R. Brian Dyer 2,* 1 Division of Natural Sciences, Oglethorpe University, Atlanta, GA 30319, USA 2 Department of Chemistry, Emory University, Atlanta, GA 30322, USA; [email protected] (C.B.); [email protected] (Q.L.); [email protected] (A.R.) * Correspondence: [email protected] (M.B.V.); [email protected] (R.B.D.); Tel.: +1-404-504-1214 (M.B.V.); +1-404-727-6637 (R.B.D.) Academic Editor: Rafik Karaman Received: 24 July 2020; Accepted: 21 August 2020; Published: 22 August 2020 Abstract: Many enzymes are known to change conformations during their catalytic cycle, but the role of these protein motions is not well understood. Escherichia coli dihydrofolate reductase (DHFR) is a small, flexible enzyme that is often used as a model system for understanding enzyme dynamics. Recently, native tryptophan fluorescence was used as a probe to study micro- to millisecond dynamics of DHFR. Yet, because DHFR has five native tryptophans, the origin of the observed conformational changes could not be assigned to a specific region within the enzyme. Here, we use DHFR mutants, each with a single tryptophan as a probe for temperature jump fluorescence spectroscopy, to further inform our understanding of DHFR dynamics. The equilibrium tryptophan fluorescence of the mutants shows that each tryptophan is in a different environment and that wild-type DHFR fluorescence is not a simple summation of all the individual tryptophan fluorescence signatures due to tryptophan–tryptophan interactions. -

Predicting Protein-Membrane Interfaces of Peripheral Membrane
bioRxiv preprint doi: https://doi.org/10.1101/2021.06.28.450157; this version posted June 29, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Predicting protein-membrane interfaces of pe- ripheral membrane proteins using ensemble machine learning Alexios Chatzigoulas1,2,* and Zoe Cournia1,* 1Biomedical Research Foundation, Academy of Athens, 4 Soranou Ephessiou, 11527 Athens, Greece, 2Depart- ment of Informatics and Telecommunications, National and Kapodistrian University of Athens, 15784 Athens, Greece *To whom correspondence should be addressed. Abstract Motivation: Abnormal protein-membrane attachment is involved in deregulated cellular pathways and in disease. Therefore, the possibility to modulate protein-membrane interactions represents a new promising therapeutic strategy for peripheral membrane proteins that have been considered so far undruggable. A major obstacle in this drug design strategy is that the membrane binding domains of peripheral membrane proteins are usually not known. The development of fast and efficient algorithms predicting the protein-membrane interface would shed light into the accessibility of membrane-protein interfaces by drug-like molecules. Results: Herein, we describe an ensemble machine learning methodology and algorithm for predicting membrane-penetrating residues. We utilize available experimental data in the literature for training 21 machine learning classifiers and a voting classifier. Evaluation of the ensemble classifier accuracy pro- duced a macro-averaged F1 score = 0.92 and an MCC = 0.84 for predicting correctly membrane-pen- etrating residues on unknown proteins of an independent test set. -

Recent Advances in the Structural Biology of the 26S Proteasome
The International Journal of Biochemistry & Cell Biology 79 (2016) 437–442 Contents lists available at ScienceDirect The International Journal of Biochemistry & Cell Biology jo urnal homepage: www.elsevier.com/locate/biocel Review article Recent advances in the structural biology of the 26S proteasome ∗ Marc Wehmer, Eri Sakata Department of Molecular Structural Biology, Max Planck institute of Biochemistry, 82152, Martinsried, Germany a r t i c l e i n f o a b s t r a c t Article history: There is growing appreciation for the fundamental role of structural dynamics in the function of macro- Received 28 June 2016 molecules. In particular, the 26S proteasome, responsible for selective protein degradation in an ATP Received in revised form 2 August 2016 dependent manner, exhibits dynamic conformational changes that enable substrate processing. Recent Accepted 3 August 2016 cryo-electron microscopy (cryo-EM) work has revealed the conformational dynamics of the 26S protea- Available online 4 August 2016 some and established the function of the different conformational states. Technological advances such as direct electron detectors and image processing algorithms allowed resolving the structure of the pro- Keywords: teasome at atomic resolution. Here we will review those studies and discuss their contribution to our 26S proteasome understanding of proteasome function. Cryoelectron microscopy © 2016 The Authors. Published by Elsevier Ltd. This is an open access article under the CC BY-NC-ND Single particle analysis Structural biology license (http://creativecommons.org/licenses/by-nc-nd/4.0/). AAA+ ATPase Contents 1. Introduction . 437 2. Structural dynamics of the 26S proteasome . 438 3. Mechanical insights into the proteasome . -

Topological Distribution of Four-A-Helix Bundles (Folding Motif/Solvent Accessibility/Helix Dipole) SCOTT R
Proc. NatI. Acad. Sci. USA Vol. 86, pp. 6592-6596, September 1989 Biochemistry Topological distribution of four-a-helix bundles (folding motif/solvent accessibility/helix dipole) SCOTT R. PRESNELL* AND FRED E. COHEN*t Departments of *Pharmaceutical Chemistry and tMedicine, University of California, San Francisco, San Francisco, CA 94143-0446 Communicated by Frederic M. Richards, June 19, 1989 ABSTRACT The four-a-helix bundle, a common struc- suggest a set of bundle structures beyond those initially tural motif in globular proteins, provides an excellent forum for reviewed by Weber and Salemme (8). Further, we will the examination of predictive constraints for protein backbone describe robust topological characterizations and categori- topology. An exhaustive examination of the Brookhaven Crys- zations of the discovered structures. In light of our catego- tallographic Protein Data Bank and other literature sources has rization scheme, the past and current topological constraints lead to the discovery of 20 putative four-a-helix bundles. used for the prediction of protein structures containing four- Application of an analytical method that examines the differ- a-helix bundles are reevaluated. ence between solvent-accessible surface areas in packed and partially unpacked bundles reduced the number of structures to 16. Angular requirements further reduced the list ofbundles METHODS to 13. In 12 of these bundles, all pairs of neighboring helices To locate putative four-a-helix bundles, two independent were oriented in an anti-parallel fashion. This distribution is in observers employed the graphical display program MIDAS accordance with structure types expected if the helix macro (11) to inspect more than 300 globular protein structures from dipole effect makes a substantial contribution to the stability of the Brookhaven Protein Data Bank (12) (November 14, 1988). -

Hydrophobic Effect, Water Structure, and Heat Capacity Changes
J. Phys. Chem. B 1997, 101, 4343-4348 4343 Hydrophobic Effect, Water Structure, and Heat Capacity Changes Kim A. Sharp* and Bhupinder Madan Department of Biochemistry & Biophysics, UniVersity of PennsylVania, 3700 Hamilton Walk, Philadelphia, PennsylVania 19104-6059 ReceiVed: January 16, 1997; In Final Form: April 1, 1997X hyd The hydration heat capacity (∆Cp ) of nine solutes of varying hydrophobicity was studied using a combination of a random network model equation of water and Monte Carlo simulations. Nonpolar solutes cause a concerted decrease in the average length and angle of the water-water hydrogen bonds in the first hydration shell, while polar and ionic solutes have the opposite effect. This is due to changes in the amounts, relative to bulk water, of two populations of hydrogen bonds: one with shorter and more linear bonds and the other with longer and more bent bonds. Heat capacity changes were calculated from these changes in water structure using a random network model equation of state. The calculated changes account for observed hyd differences in ∆Cp for the various solutes. The simulations provide a unified picture of hydrophobic and hyd polar hydration, and both a structural and thermodynamic explanation of the opposite signs of ∆Cp observed for polar and nonpolar solutes. Introduction nonpolar groups means that at higher temperatures (T > 80 °C) their insolubility results from unfavorable changes in enthalpy, In recent years analysis of heat capacity changes has become not entropy. Second, the hydration of a majority of polar and of central importance in understanding the thermodynamics of ionic solutes is also accompanied by a decrease in entropy, and protein folding, protein-protein binding, protein-nucleic acid thus by some kind of structuring of water, but in this case binding, and the hydrophobic effect.1-7 There are several hyd 7,16-20 reasons for this. -

Packing of Secondary Structures II
7.88 Lecture Notes - 5 7.24/7.88J/5.48J The Protein Folding and Human Disease Packing of Secondary Structures • Packing of Helices against sheets • Packing of sheets against sheets • Parallel • Orthogonal Table: “Amino Acid Composition of the Ten Proteins and of the Residues at the Helix to Helix Interfaces” Name Total % Total At contacts % at contacts Gly 182 9 15 4 Ala 191 9 49 12 Val 151 7 46 12 Leu 148 7 48 12 Ile 114 6 36 9 Pro 67 3 41 1 Phe 68 3 25 6 Tyr 87 6 14 4 Trp 35 2 7 2 His 45 2 18 5 ½Cys 21 1 3 1 Met 29 1 10 3 Ser 165 8 19 5 Thr 132 7 21 5 Asp 112 6 14 4 Asn 113 6 13 3 Glu 94 5 13 3 Gln 70 3 12 3 Lys 125 6 19 5 Arg 76 4 13 3 A. Factors Contributing to Stability of Correctly Folded Native State 1. Major source of stability = removal of hydrophobic side chains atoms from the solvent and burying in environment which excludes the solvent (Entropic contribution from water structure). 1 2. Formation of hydrogen bonds between buried amide and carbonyl groups is maximized 3. Retention of backbone conformations close to the minimal energies. 4. Close packing means optimal Van der Waals interactions. You have read about alpha/beta proteins in Brandon and Tooze. B. Helix to Sheet Packing Lets examine buried contacts between the helices and the sheets. First a quick review of beta sheet structure: Colored transparency: Theoretical model, not actual sheet. -

Amino Acid Preference Against Beta Sheet Through Allowing Backbone Hydration Enabled by the Presence of Cation
Amino acid preference against beta sheet through allowing backbone hydration enabled by the presence of cation John N. Sharley, University of Adelaide. arXiv 2016-10-03 [email protected] Table of Contents 1 Abstract 1 2 Introduction 2 2.1 Alpha helix preferring amino acid residues in a beta sheet 2 2.2 Cation interactions with protein backbone oxygen 3 2.3 Quantum molecular dynamics with quantum mechanical treatment of every water molecule 3 3 Methods 4 4 Results 5 4.1 Preparation 5 4.2 Experiment 1302 5 4.3 Experiment 1303 7 5 Discussion 9 5.1 HB networks of water 9 5.2 Subsequent to rupture of a transient beta sheet 9 5.3 Hofmeister effects 10 6 Conclusion 11 7 Future work 12 8 Acknowledgements 13 9 References 14 10 Appendix 1. Backbone hydration in experiment 1302 16 11 Appendix 2. Backbone hydration in experiment 1303 17 1 Abstract It is known that steric blocking by peptide sidechains of hydrogen bonding, HB, between water and peptide groups, PGs, in beta sheets accords with an amino acid intrinsic beta sheet preference [1]. The present observations with Quantum Molecular Dynamics, QMD, simulation with Quantum Mechanical, QM, treatment of every water molecule solvating a beta sheet that would be transient in nature suggest that this steric blocking is not applicable in a hydrophobic region unless a cation is present, so that the amino acid beta sheet preference due to this steric blocking is only effective in the presence of a cation. We observed backbone hydration in a polyalanine and to a lesser extent polyvaline alpha helix without a cation being present, but a cation could increase the strength of these HBs.