Structural Insights Into Binding of STAC Proteins to Voltage-Gated
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Characterization of Cecal Smooth Muscle Contraction in Laying Hens
veterinary sciences Communication Characterization of Cecal Smooth Muscle Contraction in Laying Hens Katrin Röhm 1, Martin Diener 2 , Korinna Huber 1 and Jana Seifert 1,* 1 Institute of Animal Science, University of Hohenheim, 70593 Stuttgart, Germany; [email protected] (K.R.); [email protected] (K.H.) 2 Institute of Veterinary Physiology and Biochemistry, Justus-Liebig University, 35392 Gießen, Germany; [email protected] * Correspondence: [email protected] Abstract: The ceca play an important role in the physiology of the gastrointestinal tract in chickens. Nevertheless, there is a gap of knowledge regarding the functionality of the ceca in poultry, especially with respect to physiological cecal smooth muscle contraction. The aim of the current study is the ex vivo characterization of cecal smooth muscle contraction in laying hens. Muscle strips of circular cecal smooth muscle from eleven hens are prepared to investigate their contraction ex vivo. Contraction is detected using an isometric force transducer, determining its frequency, height and intensity. Spontaneous contraction of the chicken cecal smooth muscle and the influence of buffers (calcium-free buffer and potassium-enriched buffer) and drugs (carbachol, nitroprusside, isoprenaline and Verapamil) affecting smooth muscle contraction at different levels are characterized. A decrease in smooth muscle contraction is observed when a calcium-free buffer is used. Carbachol causes an increase in smooth muscle contraction, whereas atropine inhibits contraction. Nitroprusside, isoprenaline and Verapamil result in a depression of smooth muscle contraction. In conclusion, the present results confirm a similar contraction behavior of cecal smooth muscles in laying hens as Citation: Röhm, K.; Diener, M.; shown previously in other species. -

Single-Particle Cryo-EM of the Ryanodine Receptor Channel in an Aqueous Environment
Single-particle cryo-EM of the ryanodine receptor channel Eur J Transl Myol - Basic Appl Myol 2015; 25 (1): 35-48 Single-particle cryo-EM of the ryanodine receptor channel in an aqueous environment Mariah R. Baker, Guizhen Fan and Irina I. Serysheva Department of Biochemistry and Molecular Biology, The University of Texas Medical School at Houston, 6431 Fannin Street, Houston, TX 77030, USA Abstract Ryanodine receptors (RyRs) are tetrameric ligand-gated Ca2+ release channels that are responsible for the increase of cytosolic Ca2+ concentration leading to muscle contraction. Our current understanding of RyR channel gating and regulation is greatly limited due to the lack of a high-resolution structure of the channel protein. The enormous size and unwieldy shape of Ca2+ release channels make X-ray or NMR methods difficult to apply for high-resolution structural analysis of the full-length functional channel. Single-particle electron cryo- microscopy (cryo-EM) is one of the only effective techniques for the study of such a large integral membrane protein and its molecular interactions. Despite recent developments in cryo- EM technologies and break-through single-particle cryo-EM studies of ion channels, cryospecimen preparation, particularly the presence of detergent in the buffer, remains the main impediment to obtaining atomic-resolution structures of ion channels and a multitude of other integral membrane protein complexes. In this review we will discuss properties of several detergents that have been successfully utilized in cryo-EM studies of ion channels and the emergence of the detergent alternative amphipol to stabilize ion channels for structure- function characterization. Future structural studies of challenging specimen like ion channels are likely to be facilitated by cryo-EM amenable detergents or alternative surfactants. -

Minding the Calcium Store: Ryanodine Receptor Activation As a Convergent Mechanism of PCB Toxicity
Pharmacology & Therapeutics 125 (2010) 260–285 Contents lists available at ScienceDirect Pharmacology & Therapeutics journal homepage: www.elsevier.com/locate/pharmthera Associate Editor: Carey Pope Minding the calcium store: Ryanodine receptor activation as a convergent mechanism of PCB toxicity Isaac N. Pessah ⁎, Gennady Cherednichenko, Pamela J. Lein Department of Molecular Biosciences, School of Veterinary Medicine, University of California, Davis, CA 95616, USA article info abstract Keywords: Chronic low-level polychlorinated biphenyl (PCB) exposures remain a significant public health concern since Ryanodine receptor (RyR) results from epidemiological studies indicate that PCB burden is associated with immune system Calcium-induced calcium release dysfunction, cardiovascular disease, and impairment of the developing nervous system. Of these various Calcium regulation adverse health effects, developmental neurotoxicity has emerged as a particularly vulnerable endpoint in Polychlorinated biphenyls PCB toxicity. Arguably the most pervasive biological effects of PCBs could be mediated by their ability to alter Triclosan fi 2+ Bastadins the spatial and temporal delity of Ca signals through one or more receptor-mediated processes. This Polybrominated diphenylethers review will focus on our current knowledge of the structure and function of ryanodine receptors (RyRs) in Developmental neurotoxicity muscle and nerve cells and how PCBs and related non-coplanar structures alter these functions. The Activity dependent plasticity molecular and cellular mechanisms by which non-coplanar PCBs and related structures alter local and global Ca2+ signaling properties and the possible short and long-term consequences of these perturbations on neurodevelopment and neurodegeneration are reviewed. © 2009 Elsevier Inc. All rights reserved. Contents 1. Introduction ............................................... 260 2. Ryanodine receptor macromolecular complexes: significance to polychlorinated biphenyl-mediated Ca2+ dysregulation . -

Comparative Genomic Analysis of Integral Membrane Transport Proteins in Ciliates
UC San Diego UC San Diego Previously Published Works Title Comparative genomic analysis of integral membrane transport proteins in ciliates. Permalink https://escholarship.org/uc/item/3g98s19z Journal The Journal of eukaryotic microbiology, 62(2) ISSN 1066-5234 Authors Kumar, Ujjwal Saier, Milton H Publication Date 2015-03-01 DOI 10.1111/jeu.12156 Peer reviewed eScholarship.org Powered by the California Digital Library University of California The Journal of Published by the International Society of Eukaryotic Microbiology Protistologists Journal of Eukaryotic Microbiology ISSN 1066-5234 ORIGINAL ARTICLE Comparative Genomic Analysis of Integral Membrane Transport Proteins in Ciliates Ujjwal Kumar & Milton H. Saier Jr Division of Biological Sciences, University of California at San Diego, La Jolla, California Keywords ABSTRACT Channels; evolution; genome analyses; secondary carriers. Integral membrane transport proteins homologous to those found in the Transporter Classification Database (TCDB; www.tcdb.org) were identified and Correspondence bioinformatically characterized by transporter class, family, and substrate speci- M. H. Saier Jr, Division of Biological ficity in three ciliates, Paramecium tetraurelia (Para), Tetrahymena thermophila Sciences, University of California at San (Tetra), and Ichthyophthirius multifiliis (Ich). In these three organisms, 1,326 of Diego, La Jolla, CA 92093-0116, USA 39,600 proteins (3.4%), 1,017 of 24,800 proteins (4.2%), and 504 out of 8,100 Telephone number: +858-534-4084; proteins (6.2%) integral membrane transport proteins were identified, respec- FAX number: +858-534-7108; tively. Thus, an inverse relationship was observed between the % transporters e-mail: [email protected] identified and the number of total proteins per genome reported. -

Back-To-Basics: the Intricacies of Muscle Contraction
Back-to- MIOTA Basics: The CONFERENCE OCTOBER 11, Intricacies 2019 CHERI RAMIREZ, MS, of Muscle OTRL Contraction OBJECTIVES: 1.Review the anatomical structure of a skeletal muscle. 2.Review and understand the process and relationship between skeletal muscle contraction with the vital components of the nervous system, endocrine system, and skeletal system. 3.Review the basic similarities and differences between skeletal muscle tissue, smooth muscle tissue, and cardiac muscle tissue. 4.Review the names, locations, origins, and insertions of the skeletal muscles found in the human body. 5.Apply the information learned to enhance clinical practice and understanding of the intricacies and complexity of the skeletal muscle system. 6.Apply the information learned to further educate clients on the importance of skeletal muscle movement, posture, and coordination in the process of rehabilitation, healing, and functional return. 1. Epithelial Four Basic Tissue Categories 2. Muscle 3. Nervous 4. Connective A. Loose Connective B. Bone C. Cartilage D. Blood Introduction There are 3 types of muscle tissue in the muscular system: . Skeletal muscle: Attached to bones of skeleton. Voluntary. Striated. Tubular shape. Cardiac muscle: Makes up most of the wall of the heart. Involuntary. Striated with intercalated discs. Branched shape. Smooth muscle: Found in walls of internal organs and walls of vascular system. Involuntary. Non-striated. Spindle shape. 4 Structure of a Skeletal Muscle Skeletal Muscles: Skeletal muscles are composed of: • Skeletal muscle tissue • Nervous tissue • Blood • Connective tissues 5 Connective Tissue Coverings Connective tissue coverings over skeletal muscles: .Fascia .Tendons .Aponeuroses 6 Fascia: Definition: Layers of dense connective tissue that separates muscle from adjacent muscles, by surrounding each muscle belly. -

Snapshot: Mammalian TRP Channels David E
SnapShot: Mammalian TRP Channels David E. Clapham HHMI, Children’s Hospital, Department of Neurobiology, Harvard Medical School, Boston, MA 02115, USA TRP Activators Inhibitors Putative Interacting Proteins Proposed Functions Activation potentiated by PLC pathways Gd, La TRPC4, TRPC5, calmodulin, TRPC3, Homodimer is a purported stretch-sensitive ion channel; form C1 TRPP1, IP3Rs, caveolin-1, PMCA heteromeric ion channels with TRPC4 or TRPC5 in neurons -/- Pheromone receptor mechanism? Calmodulin, IP3R3, Enkurin, TRPC6 TRPC2 mice respond abnormally to urine-based olfactory C2 cues; pheromone sensing 2+ Diacylglycerol, [Ca ]I, activation potentiated BTP2, flufenamate, Gd, La TRPC1, calmodulin, PLCβ, PLCγ, IP3R, Potential role in vasoregulation and airway regulation C3 by PLC pathways RyR, SERCA, caveolin-1, αSNAP, NCX1 La (100 µM), calmidazolium, activation [Ca2+] , 2-APB, niflumic acid, TRPC1, TRPC5, calmodulin, PLCβ, TRPC4-/- mice have abnormalities in endothelial-based vessel C4 i potentiated by PLC pathways DIDS, La (mM) NHERF1, IP3R permeability La (100 µM), activation potentiated by PLC 2-APB, flufenamate, La (mM) TRPC1, TRPC4, calmodulin, PLCβ, No phenotype yet reported in TRPC5-/- mice; potentially C5 pathways, nitric oxide NHERF1/2, ZO-1, IP3R regulates growth cones and neurite extension 2+ Diacylglycerol, [Ca ]I, 20-HETE, activation 2-APB, amiloride, Cd, La, Gd Calmodulin, TRPC3, TRPC7, FKBP12 Missense mutation in human focal segmental glomerulo- C6 potentiated by PLC pathways sclerosis (FSGS); abnormal vasoregulation in TRPC6-/- -

Ca Signaling in Cardiac Fibroblasts and Fibrosis-Associated Heart
Journal of Cardiovascular Development and Disease Review Ca2+ Signaling in Cardiac Fibroblasts and Fibrosis-Associated Heart Diseases Jianlin Feng 1, Maria K. Armillei 1, Albert S. Yu 1, Bruce T. Liang 1, Loren W. Runnels 2,* and Lixia Yue 1,* 1 Calhoun Cardiology Center, Department of Cell Biology, University of Connecticut Health Center, Farmington, CT 06030, USA; [email protected] (J.F.); [email protected] (M.K.A.); [email protected] (A.S.Y.); [email protected] (B.T.L.) 2 Department of Pharmacology, Rutgers, Robert Wood Johnson Medical School, Piscataway, NJ 08854, USA * Correspondence: [email protected] (L.W.R.); [email protected] (L.Y.) Received: 11 August 2019; Accepted: 18 September 2019; Published: 23 September 2019 Abstract: Cardiac fibrosis is the excessive deposition of extracellular matrix proteins by cardiac fibroblasts and myofibroblasts, and is a hallmark feature of most heart diseases, including arrhythmia, hypertrophy, and heart failure. This maladaptive process occurs in response to a variety of stimuli, including myocardial injury, inflammation, and mechanical overload. There are multiple signaling pathways and various cell types that influence the fibrogenesis cascade. Fibroblasts and myofibroblasts are central effectors. Although it is clear that Ca2+ signaling plays a vital role in this pathological process, what contributes to Ca2+ signaling in fibroblasts and myofibroblasts is still not wholly understood, chiefly because of the large and diverse number of receptors, transporters, and ion channels that influence intracellular Ca2+ signaling. Intracellular Ca2+ signals are generated by Ca2+ release from intracellular Ca2+ stores and by Ca2+ entry through a multitude of Ca2+-permeable ion channels in the plasma membrane. -

Muscle Physiology Dr
Muscle Physiology Dr. Ebneshahidi Copyright © 2004 Pearson Education, Inc., publishing as Benjamin Cummings Skeletal Muscle Figure 9.2 (a) Copyright © 2004 Pearson Education, Inc., publishing as Benjamin Cummings Functions of the muscular system . 1. Locomotion . 2. Vasoconstriction and vasodilatation- constriction and dilation of blood vessel Walls are the results of smooth muscle contraction. 3. Peristalsis – wavelike motion along the digestive tract is produced by the Smooth muscle. 4. Cardiac motion . 5. Posture maintenance- contraction of skeletal muscles maintains body posture and muscle tone. 6. Heat generation – about 75% of ATP energy used in muscle contraction is released as heat. Copyright. © 2004 Pearson Education, Inc., publishing as Benjamin Cummings . Striation: only present in skeletal and cardiac muscles. Absent in smooth muscle. Nucleus: smooth and cardiac muscles are uninculcated (one nucleus per cell), skeletal muscle is multinucleated (several nuclei per cell ). Transverse tubule ( T tubule ): well developed in skeletal and cardiac muscles to transport calcium. Absent in smooth muscle. Intercalated disk: specialized intercellular junction that only occurs in cardiac muscle. Control: skeletal muscle is always under voluntary control‚ with some exceptions ( the tongue and pili arrector muscles in the dermis). smooth and cardiac muscles are under involuntary control. Copyright © 2004 Pearson Education, Inc., publishing as Benjamin Cummings Innervation: motor unit . a) a motor nerve and a myofibril from a neuromuscular junction where gap (called synapse) occurs between the two structures. at the end of motor nerve‚ neurotransmitter (i.e. acetylcholine) is stored in synaptic vesicles which will release the neurotransmitter using exocytosis upon the stimulation of a nerve impulse. Across the synapse the surface the of myofibril contains receptors that can bind with the neurotransmitter. -

Preclinical Model Systems of Ryanodine Receptor 1-Related Myopathies and Malignant Hyperthermia
Lawal et al. Orphanet Journal of Rare Diseases (2020) 15:113 https://doi.org/10.1186/s13023-020-01384-x REVIEW Open Access Preclinical model systems of ryanodine receptor 1-related myopathies and malignant hyperthermia: a comprehensive scoping review of works published 1990– 2019 Tokunbor A. Lawal1, Emily S. Wires2, Nancy L. Terry3, James J. Dowling4 and Joshua J. Todd1* Abstract Background: Pathogenic variations in the gene encoding the skeletal muscle ryanodine receptor (RyR1) are associated with malignant hyperthermia (MH) susceptibility, a life-threatening hypermetabolic condition and RYR1- related myopathies (RYR1-RM), a spectrum of rare neuromuscular disorders. In RYR1-RM, intracellular calcium dysregulation, post-translational modifications, and decreased protein expression lead to a heterogenous clinical presentation including proximal muscle weakness, contractures, scoliosis, respiratory insufficiency, and ophthalmoplegia. Preclinical model systems of RYR1-RM and MH have been developed to better understand underlying pathomechanisms and test potential therapeutics. Methods: We conducted a comprehensive scoping review of scientific literature pertaining to RYR1-RM and MH preclinical model systems in accordance with the PRISMA Scoping Reviews Checklist and the framework proposed by Arksey and O’Malley. Two major electronic databases (PubMed and EMBASE) were searched without language restriction for articles and abstracts published between January 1, 1990 and July 3, 2019. Results: Our search yielded 5049 publications from which 262 were included in this review. A majority of variants tested in RYR1 preclinical models were localized to established MH/central core disease (MH/CCD) hot spots. A total of 250 unique RYR1 variations were reported in human/rodent/porcine models with 95% being missense substitutions. -
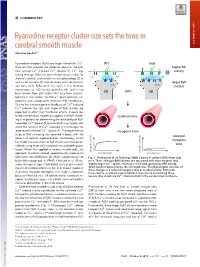
Ryanodine Receptor Cluster Size Sets the Tone in Cerebral Smooth Muscle
COMMENTARY Ryanodine receptor cluster size sets the tone in cerebral smooth muscle COMMENTARY Christian Soellera,1 + Ryanodine receptors (RyRs) are large intracellular Ca2 WT mdx channels that provide the molecular basis of the pro- K+ K+ higher BK + + + cess termed Ca2 -induced Ca2 release (1). Ca2 sig- activity naling through RyRs has been shown to be critical for CaC BK BK skeletal, cardiac, and smooth muscle physiology (2) as well as for neurons (3) and secretory cells like pancre- larger RyR atic beta cells. RyRs were first seen in the electron RyRs RyRs clusters microscope as ∼30-nm-size particles (4), and it had SMCs been known from EM studies that they form clusters, SR SR typically in the narrow “junctional” space between sar- colemma and sarcoplasmic reticulum (SR) membranes. + Due to the inherent positive feedback of Ca2 -induced + Ca2 release, the size and shape of RyR clusters are expected to affect their functional activity. Indeed, de- SMCs SMCs tailed mathematical modeling suggests that RyR cluster- cerebral artery ing is important for determining the excitability of RyR- + mediated Ca2 release (5, 6) and could, in principle, also + affect the amount of Ca2 released in microscopic re- 2+ lease events termed Ca sparks (7). The experimental myogenic tone study of RyR clustering has received a boost with the reduced advent of optical superresolution microscopy, which myogenic has made the assessment of RyR cluster size more acces- sible by using essentially standard immunolabeling pro- tone tocols. When first applied in cardiac muscle cells, this myogenic tone (%) tone myogenic approach revealed a broad, approximately exponential vessel pressure (%) tone myogenic vessel pressure RyR cluster size distribution (8), which had previously not Fig. -

Oxidative Stress Induces Stem Cell Proliferation Via TRPA1/Ryr
RESEARCH ARTICLE Oxidative stress induces stem cell proliferation via TRPA1/RyR-mediated Ca2+ signaling in the Drosophila midgut Chiwei Xu1, Junjie Luo2,3, Li He1, Craig Montell2,3, Norbert Perrimon1,4* 1Department of Genetics, Harvard Medical School, Boston, United States; 2Department of Molecular, Cellular and Developmental Biology, University of California Santa Barbara, Santa Barbara, United States; 3Neuroscience Research Institute, University of California, Santa Barbara, Santa Barbara, United States; 4Howard Hughes Medical Institute, Harvard Medical School, Boston, United States Abstract Precise regulation of stem cell activity is crucial for tissue homeostasis and necessary to prevent overproliferation. In the Drosophila adult gut, high levels of reactive oxygen species (ROS) has been detected with different types of tissue damage, and oxidative stress has been shown to be both necessary and sufficient to trigger intestinal stem cell (ISC) proliferation. However, the connection between oxidative stress and mitogenic signals remains obscure. In a screen for genes required for ISC proliferation in response to oxidative stress, we identified two regulators of cytosolic Ca2+ levels, transient receptor potential A1 (TRPA1) and ryanodine receptor (RyR). Characterization of TRPA1 and RyR demonstrates that Ca2+ signaling is required for oxidative stress-induced activation of the Ras/MAPK pathway, which in turns drives ISC proliferation. Our findings provide a link between redox regulation and Ca2+ signaling and reveal a novel mechanism by which ISCs detect stress signals. DOI: 10.7554/eLife.22441.001 *For correspondence: perrimon@ Introduction receptor.med.harvard.edu Multipotent intestinal stem cells (ISCs) are responsible for tissue homeostasis in the adult Drosophila Competing interests: The midgut (Jiang and Edgar, 2011; Micchelli and Perrimon, 2006; Ohlstein and Spradling, 2006). -

Original Article Unbalanced Upregulation of Ryanodine Receptor 2 Plays a Particular Role in Early Development of Daunorubicin Cardiomyopathy
Am J Transl Res 2015;7(7):1280-1294 www.ajtr.org /ISSN:1943-8141/AJTR0010909 Original Article Unbalanced upregulation of ryanodine receptor 2 plays a particular role in early development of daunorubicin cardiomyopathy Dana Kucerova1,2, Gabriel Doka1, Peter Kruzliak3, Katarina Turcekova1, Jana Kmecova1, Zuzana Brnoliakova4, Jan Kyselovic1, Uwe Kirchhefer2, Frank U Müller2, Peter Krenek1, Peter Boknik2, Jan Klimas1 1Department of Pharmacology and Toxicology, Faculty of Pharmacy, Comenius University, Bratislava, Slovak Re- public; 2Institut für Pharmakologie und Toxikologie, Universitätsklinikum Münster, Münster, Germany; 3Internation- al Clinical Research Center, St. Anne’s University Hospital and Masaryk University, Brno, Czech Republic; 4Institute of Experimental Pharmacology, Slovak Academy of Sciences, Bratislava, Slovak Republic Received May 31, 2015; Accepted July 14, 2015; Epub July 15, 2015; Published July 30, 2015 Abstract: Calcium release channel on the sarcoplasmic reticulum of cardiomyocytes (ryanodine receptor type 2, RyR2) plays a critical role in the regulation of calcium and was identified as a crucial factor for development of chronic anthracycline cardiomyopathy. Its early stages are less well described although these determine the later development. Hence, we tested the effect of repeated, short-term anthracycline (daunorubicin) administration on cardiac performance, cardiomyocyte function and accompanied changes in calcium regulating proteins expression. Ten-twelve weeks old male Wistar rats were administered with 6 doses of daunorubicin (DAU, 3 mg/kg, i.p., every 48 h), controls (CON) received vehicle. Left ventricular function (left ventricular pressure, LVP; rate of pressure de- velopment, +dP/dt and decline, -dP/dt) was measured using left ventricular catheterization under tribromethanol anaesthesia (15 ml/kg b.w.).