Lecture Notes on Intermediate Thermodynamics
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
![Arxiv:2008.06405V1 [Physics.Ed-Ph] 14 Aug 2020 Fig.1 Shows Four Classical Cycles: (A) Carnot Cycle, (B) Stirling Cycle, (C) Otto Cycle and (D) Diesel Cycle](https://docslib.b-cdn.net/cover/0049/arxiv-2008-06405v1-physics-ed-ph-14-aug-2020-fig-1-shows-four-classical-cycles-a-carnot-cycle-b-stirling-cycle-c-otto-cycle-and-d-diesel-cycle-70049.webp)
Arxiv:2008.06405V1 [Physics.Ed-Ph] 14 Aug 2020 Fig.1 Shows Four Classical Cycles: (A) Carnot Cycle, (B) Stirling Cycle, (C) Otto Cycle and (D) Diesel Cycle
Investigating student understanding of heat engine: a case study of Stirling engine Lilin Zhu1 and Gang Xiang1, ∗ 1Department of Physics, Sichuan University, Chengdu 610064, China (Dated: August 17, 2020) We report on the study of student difficulties regarding heat engine in the context of Stirling cycle within upper-division undergraduate thermal physics course. An in-class test about a Stirling engine with a regenerator was taken by three classes, and the students were asked to perform one of the most basic activities—calculate the efficiency of the heat engine. Our data suggest that quite a few students have not developed a robust conceptual understanding of basic engineering knowledge of the heat engine, including the function of the regenerator and the influence of piston movements on the heat and work involved in the engine. Most notably, although the science error ratios of the three classes were similar (∼10%), the engineering error ratios of the three classes were high (above 50%), and the class that was given a simple tutorial of engineering knowledge of heat engine exhibited significantly smaller engineering error ratio by about 20% than the other two classes. In addition, both the written answers and post-test interviews show that most of the students can only associate Carnot’s theorem with Carnot cycle, but not with other reversible cycles working between two heat reservoirs, probably because no enough cycles except Carnot cycle were covered in the traditional Thermodynamics textbook. Our results suggest that both scientific and engineering knowledge are important and should be included in instructional approaches, especially in the Thermodynamics course taught in the countries and regions with a tradition of not paying much attention to experimental education or engineering training. -

Equilibrium Thermodynamics
Equilibrium Thermodynamics Instructor: - Clare Yu (e-mail [email protected], phone: 824-6216) - office hours: Wed from 2:30 pm to 3:30 pm in Rowland Hall 210E Textbooks: - primary: Herbert Callen “Thermodynamics and an Introduction to Thermostatistics” - secondary: Frederick Reif “Statistical and Thermal Physics” - secondary: Kittel and Kroemer “Thermal Physics” - secondary: Enrico Fermi “Thermodynamics” Grading: - weekly homework: 25% - discussion problems: 10% - midterm exam: 30% - final exam: 35% Equilibrium Thermodynamics Material Covered: Equilibrium thermodynamics, phase transitions, critical phenomena (~ 10 first chapters of Callen’s textbook) Homework: - Homework assignments posted on course website Exams: - One midterm, 80 minutes, Tuesday, May 8 - Final, 2 hours, Tuesday, June 12, 10:30 am - 12:20 pm - All exams are in this room 210M RH Course website is at http://eiffel.ps.uci.edu/cyu/p115B/class.html The Subject of Thermodynamics Thermodynamics describes average properties of macroscopic matter in equilibrium. - Macroscopic matter: large objects that consist of many atoms and molecules. - Average properties: properties (such as volume, pressure, temperature etc) that do not depend on the detailed positions and velocities of atoms and molecules of macroscopic matter. Such quantities are called thermodynamic coordinates, variables or parameters. - Equilibrium: state of a macroscopic system in which all average properties do not change with time. (System is not driven by external driving force.) Why Study Thermodynamics ? - Thermodynamics predicts that the average macroscopic properties of a system in equilibrium are not independent from each other. Therefore, if we measure a subset of these properties, we can calculate the rest of them using thermodynamic relations. - Thermodynamics not only gives the exact description of the state of equilibrium but also provides an approximate description (to a very high degree of precision!) of relatively slow processes. -
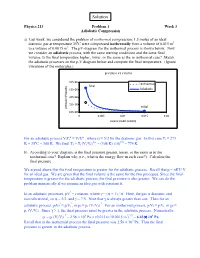
3-1 Adiabatic Compression
Solution Physics 213 Problem 1 Week 3 Adiabatic Compression a) Last week, we considered the problem of isothermal compression: 1.5 moles of an ideal diatomic gas at temperature 35oC were compressed isothermally from a volume of 0.015 m3 to a volume of 0.0015 m3. The pV-diagram for the isothermal process is shown below. Now we consider an adiabatic process, with the same starting conditions and the same final volume. Is the final temperature higher, lower, or the same as the in isothermal case? Sketch the adiabatic processes on the p-V diagram below and compute the final temperature. (Ignore vibrations of the molecules.) α α For an adiabatic process ViTi = VfTf , where α = 5/2 for the diatomic gas. In this case Ti = 273 o 1/α 2/5 K + 35 C = 308 K. We find Tf = Ti (Vi/Vf) = (308 K) (10) = 774 K. b) According to your diagram, is the final pressure greater, lesser, or the same as in the isothermal case? Explain why (i.e., what is the energy flow in each case?). Calculate the final pressure. We argued above that the final temperature is greater for the adiabatic process. Recall that p = nRT/ V for an ideal gas. We are given that the final volume is the same for the two processes. Since the final temperature is greater for the adiabatic process, the final pressure is also greater. We can do the problem numerically if we assume an idea gas with constant α. γ In an adiabatic processes, pV = constant, where γ = (α + 1) / α. -

Transcritical Pressure Organic Rankine Cycle (ORC) Analysis Based on the Integrated-Average Temperature Difference in Evaporators
Applied Thermal Engineering 88 (2015) 2e13 Contents lists available at ScienceDirect Applied Thermal Engineering journal homepage: www.elsevier.com/locate/apthermeng Research paper Transcritical pressure Organic Rankine Cycle (ORC) analysis based on the integrated-average temperature difference in evaporators * Chao Yu, Jinliang Xu , Yasong Sun The Beijing Key Laboratory of Multiphase Flow and Heat Transfer for Low Grade Energy Utilizations, North China Electric Power University, Beijing 102206, PR China article info abstract Article history: Integrated-average temperature difference (DTave) was proposed to connect with exergy destruction (Ieva) Received 24 June 2014 in heat exchangers. Theoretical expressions were developed for DTave and Ieva. Based on transcritical Received in revised form pressure ORCs, evaporators were theoretically studied regarding DTave. An exact linear relationship be- 12 October 2014 tween DT and I was identified. The increased specific heats versus temperatures for organic fluid Accepted 11 November 2014 ave eva protruded its TeQ curve to decrease DT . Meanwhile, the decreased specific heats concaved its TeQ Available online 20 November 2014 ave curve to raise DTave. Organic fluid in the evaporator undergoes a protruded TeQ curve and a concaved T eQ curve, interfaced at the pseudo-critical temperature point. Elongating the specific heat increment Keywords: fi Organic Rankine Cycle section and shortening the speci c heat decrease section improved the cycle performance. Thus, the fi Integrated-average temperature difference system thermal and exergy ef ciencies were increased by increasing critical temperatures for 25 organic Exergy destruction fluids. Wet fluids had larger thermal and exergy efficiencies than dry fluids, due to the fact that wet fluids Thermal match shortened the superheated vapor flow section in condensers. -

Entropy: Ideal Gas Processes
Chapter 19: The Kinec Theory of Gases Thermodynamics = macroscopic picture Gases micro -> macro picture One mole is the number of atoms in 12 g sample Avogadro’s Number of carbon-12 23 -1 C(12)—6 protrons, 6 neutrons and 6 electrons NA=6.02 x 10 mol 12 atomic units of mass assuming mP=mn Another way to do this is to know the mass of one molecule: then So the number of moles n is given by M n=N/N sample A N = N A mmole−mass € Ideal Gas Law Ideal Gases, Ideal Gas Law It was found experimentally that if 1 mole of any gas is placed in containers that have the same volume V and are kept at the same temperature T, approximately all have the same pressure p. The small differences in pressure disappear if lower gas densities are used. Further experiments showed that all low-density gases obey the equation pV = nRT. Here R = 8.31 K/mol ⋅ K and is known as the "gas constant." The equation itself is known as the "ideal gas law." The constant R can be expressed -23 as R = kNA . Here k is called the Boltzmann constant and is equal to 1.38 × 10 J/K. N If we substitute R as well as n = in the ideal gas law we get the equivalent form: NA pV = NkT. Here N is the number of molecules in the gas. The behavior of all real gases approaches that of an ideal gas at low enough densities. Low densitiens m= enumberans tha oft t hemoles gas molecul es are fa Nr e=nough number apa ofr tparticles that the y do not interact with one another, but only with the walls of the gas container. -

Math Background for Thermodynamics ∑
MATH BACKGROUND FOR THERMODYNAMICS A. Partial Derivatives and Total Differentials Partial Derivatives Given a function f(x1,x2,...,xm) of m independent variables, the partial derivative ∂ f of f with respect to x , holding the other m-1 independent variables constant, , is defined by i ∂ xi xj≠i ∂ f fx( , x ,..., x+ ∆ x ,..., x )− fx ( , x ,..., x ,..., x ) = 12ii m 12 i m ∂ lim ∆ xi x →∆ 0 xi xj≠i i nRT Example: If p(n,V,T) = , V ∂ p RT ∂ p nRT ∂ p nR = = − = ∂ n V ∂V 2 ∂T V VT,, nTV nV , Total Differentials Given a function f(x1,x2,...,xm) of m independent variables, the total differential of f, df, is defined by m ∂ f df = ∑ dx ∂ i i=1 xi xji≠ ∂ f ∂ f ∂ f = dx + dx + ... + dx , ∂ 1 ∂ 2 ∂ m x1 x2 xm xx2131,...,mm xxx , ,..., xx ,..., m-1 where dxi is an infinitesimally small but arbitrary change in the variable xi. nRT Example: For p(n,V,T) = , V ∂ p ∂ p ∂ p dp = dn + dV + dT ∂ n ∂ V ∂ T VT,,, nT nV RT nRT nR = dn − dV + dT V V 2 V B. Some Useful Properties of Partial Derivatives 1. The order of differentiation in mixed second derivatives is immaterial; e.g., for a function f(x,y), ∂ ∂ f ∂ ∂ f ∂ 22f ∂ f = or = ∂ y ∂ xx ∂ ∂ y ∂∂yx ∂∂xy y x x y 2 in the commonly used short-hand notation. (This relation can be shown to follow from the definition of partial derivatives.) 2. Given a function f(x,y): ∂ y 1 a. = etc. ∂ f ∂ f x ∂ y x ∂ f ∂ y ∂ x b. -

Theoretical Study of the Flow in a Fluid Damper Containing High Viscosity
Theoretical study of the flow in a fluid damper containing high viscosity silicone oil: effects of shear-thinning and viscoelasticity Alexandros Syrakosa,∗, Yannis Dimakopoulosa, John Tsamopoulosa aLaboratory of Fluid Mechanics and Rheology, Department of Chemical Engineering, University of Patras, 26500 Patras, Greece Abstract The flow inside a fluid damper where a piston reciprocates sinusoidally inside an outer casing containing high-viscosity silicone oil is simulated using a Finite Volume method, at various excitation frequencies. The oil is modelled by the Carreau-Yasuda (CY) and Phan-Thien & Tanner (PTT) constitutive equations. Both models account for shear-thinning but only the PTT model accounts for elasticity. The CY and other gener- alised Newtonian models have been previously used in theoretical studies of fluid dampers, but the present study is the first to perform full two-dimensional (axisymmetric) simulations employing a viscoelastic con- stitutive equation. It is found that the CY and PTT predictions are similar when the excitation frequency is low, but at medium and higher frequencies the CY model fails to describe important phenomena that are predicted by the PTT model and observed in experimental studies found in the literature, such as the hysteresis of the force-displacement and force-velocity loops. Elastic effects are quantified by applying a decomposition of the damper force into elastic and viscous components, inspired from LAOS (Large Am- plitude Oscillatory Shear) theory. The CY model also overestimates the damper force relative to the PTT, because it underpredicts the flow development length inside the piston-cylinder gap. It is thus concluded that (a) fluid elasticity must be accounted for and (b) theoretical approaches that rely on the assumption of one-dimensional flow in the piston-cylinder gap are of limited accuracy, even if they account for fluid viscoelasticity. -

Geometry of Thermodynamic Processes
Article Geometry of Thermodynamic Processes Arjan van der Schaft 1, and Bernhard Maschke 2 1 Bernoulli Institute for Mathematics, Computer Science and Artificial Intelligence, Jan C. Willems Center for Systems and Control, University of Groningen, the Netherlands; [email protected] 2 Univ. Lyon 1, Université Claude Bernard Lyon 1, CNRS, LAGEP UMR 5007, Villeurbanne, France; [email protected] * Correspondence: [email protected]; Tel.: +31-50-3633731 Received: date; Accepted: date; Published: date Abstract: Since the 1970s contact geometry has been recognized as an appropriate framework for the geometric formulation of the state properties of thermodynamic systems, without, however, addressing the formulation of non-equilibrium thermodynamic processes. In [3] it was shown how the symplectization of contact manifolds provides a new vantage point; enabling, among others, to switch between the energy and entropy representations of a thermodynamic system. In the present paper this is continued towards the global geometric definition of a degenerate Riemannian metric on the homogeneous Lagrangian submanifold describing the state properties, which is overarching the locally defined metrics of Weinhold and Ruppeiner. Next, a geometric formulation is given of non-equilibrium thermodynamic processes, in terms of Hamiltonian dynamics defined by Hamiltonian functions that are homogeneous of degree one in the co-extensive variables and zero on the homogeneous Lagrangian submanifold. The correspondence between objects in contact geometry and their homogeneous counterparts in symplectic geometry, as already largely present in the literature [2,18], appears to be elegant and effective. This culminates in the definition of port-thermodynamic systems, and the formulation of interconnection ports. -

IB Questionbank
Topic 3 Past Paper [94 marks] This question is about thermal energy transfer. A hot piece of iron is placed into a container of cold water. After a time the iron and water reach thermal equilibrium. The heat capacity of the container is negligible. specific heat capacity. [2 marks] 1a. Define Markscheme the energy required to change the temperature (of a substance) by 1K/°C/unit degree; of mass 1 kg / per unit mass; [5 marks] 1b. The following data are available. Mass of water = 0.35 kg Mass of iron = 0.58 kg Specific heat capacity of water = 4200 J kg–1K–1 Initial temperature of water = 20°C Final temperature of water = 44°C Initial temperature of iron = 180°C (i) Determine the specific heat capacity of iron. (ii) Explain why the value calculated in (b)(i) is likely to be different from the accepted value. Markscheme (i) use of mcΔT; 0.58×c×[180-44]=0.35×4200×[44-20]; c=447Jkg-1K-1≈450Jkg-1K-1; (ii) energy would be given off to surroundings/environment / energy would be absorbed by container / energy would be given off through vaporization of water; hence final temperature would be less; hence measured value of (specific) heat capacity (of iron) would be higher; This question is in two parts. Part 1 is about ideal gases and specific heat capacity. Part 2 is about simple harmonic motion and waves. Part 1 Ideal gases and specific heat capacity State assumptions of the kinetic model of an ideal gas. [2 marks] 2a. two Markscheme point molecules / negligible volume; no forces between molecules except during contact; motion/distribution is random; elastic collisions / no energy lost; obey Newton’s laws of motion; collision in zero time; gravity is ignored; [4 marks] 2b. -

6CCP3212 Statistical Mechanics Homework 1
6CCP3212 Statistical Mechanics Homework 1 Lecturer: Dr. Eugene A. Lim 2018-19 Year 3 Semester 1 https://nms.kcl.ac.uk/eugene.lim/teach/statmech/sm.html 1) (i) For the following differentials with α and β non-zero real constants, which are exact and which are inexact? Integrate the equation if it is exact. (a) x dG = αdx + β dy (1) y (b) α dG = dx + βdy (2) x (c) x2 dG = (x + y)dx + dy (3) 2 (ii) Show that the work done on the system at pressure P d¯W = −P dV (4) where dV is the change in volume is an inexact differential by showing that there exists no possible function of state for W (P; V ). (iii) Consider the differential dF = (x2 − y)dx + xdy : (5) (a) Show that this is not an exact differential. And hence integrate this equation in two different straight paths from (1; 1) ! (2; 2) and from (1; 1) ! (1; 2) ! (2; 2), where (x; y) indicates the locations. Compare the results { are they identical? (b) Define a new differential with dF y 1 dG ≡ = 1 − dx + dy : (6) x2 x2 x Show that dG is exact, and find G(x; y). 2) This problem asks you to derive some derivative identities of a system with three variables x, y and z, with a single constraint x(y; z). This kind of system is central to thermodynamics as we often use three state variables P , V and T , with an equation of state P (V; T ) (i.e. the constraint) to describe a system. -

179 Design of a Hydraulic Damper for Heavy Machinery
ANALELE UNIVERSIT ĂłII “EFTIMIE MURGU” RE ŞIłA ANUL XVIII, NR. 1, 2011, ISSN 1453 - 7397 Emil Zaev, Gerhard Rath, Hubert Kargl Design of a Hydraulic Damper for Heavy Machinery A hydraulic unit consisting of an accumulator as energy storage element and an orifice providing friction was designed to damp oscillations of a machine during operation. In the first step, a model for the gas spring was developed from the ideal gas laws for the dimensioning the elements. To model the gas process with a graphical simulation tool it is necessary to find a form of the gas law which can be integrated with a numerical solver, such as Tustin, Runge-Kutta, or other. For simulating the working condition, the model was refined using the van der Waals equations for real gas. A unified model representation was found to be applied for any arbitrary state change. Verifications were made with the help of special state changes, adiabatic and isothermal. After determining the dimensional parameters, which are the accumulator capacity and the orifice size, the operational and the limiting parameters were to be found. The working process of a damper includes the gas pre-charging to a predefined pressure, the nearly isothermal static loading process, and the adiabatic change during the dynamic operation. Keywords : Vibration damping, hydraulic damping, gas laws 1. Introduction Mining Machines, like the ALPINE Miner of the Sandvik company in Fig. 1, are subject to severe vibrations during their operation. Measures for damping are re- quired. Goal of this project was to investigate the potential of damping oscillation using the hydraulic cylinders of the support jacks (Fig. -

Rankine Cycle Definitions
Rankine Cycle Reading Problems 11.1 ! 11.7 11.29, 11.36, 11.43, 11.47, 11.52, 11.55, 11.58, 11.74 Definitions • working fluid is alternately vaporized and condensed as it recirculates in a closed cycle • the standard vapour cycle that excludes internal irreversibilities is called the Ideal Rankine Cycle Analyze the Process Device 1st Law Balance Boiler h2 + qH = h3 ) qH = h3 − h2 (in) Turbine h3 = h4 + wT ) wT = h3 − h4 (out) Condenser h4 = h1 + qL ) qL = h4 − h1 (out) Pump h1 + wP = h2 ) wP = h2 − h1 (in) 1 The Rankine efficiency is net work output ηR = heat supplied to the boiler (h − h ) + (h − h ) = 3 4 1 2 (h3 − h2) Effects of Boiler and Condenser Pressure We know the efficiency is proportional to T η / 1 − L TH The question is ! how do we increase efficiency ) TL # and/or TH ". 1. INCREASED BOILER PRESSURE: • an increase in boiler pressure results in a higher TH for the same TL, therefore η ". • but 40 has a lower quality than 4 – wetter steam at the turbine exhaust – results in cavitation of the turbine blades – η # plus " maintenance • quality should be > 80 − 90% at the turbine exhaust 2 2. LOWER TL: • we are generally limited by the T ER (lake, river, etc.) eg. lake @ 15 ◦C + ∆T = 10 ◦C = 25 ◦C | {z } resistance to HT ) Psat = 3:169 kP a. • this is why we have a condenser – the pressure at the exit of the turbine can be less than atmospheric pressure 3. INCREASED TH BY ADDING SUPERHEAT: • the average temperature at which heat is supplied in the boiler can be increased by superheating the steam – dry saturated steam from the boiler is passed through a second bank of smaller bore tubes within the boiler until the steam reaches the required temperature – The value of T H , the mean temperature at which heat is added, increases, while TL remains constant.