Sulfadimethoxine Analysis in Channel Catfish: a Model Drug Residue Monitoring Program for Aquaculture
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Dietary Exposure Assessment of Veterinary Antibiotics in Pork Meat on Children and Adolescents in Cyprus
foods Article Dietary Exposure Assessment of Veterinary Antibiotics in Pork Meat on Children and Adolescents in Cyprus Demetra Kyriakides 1,2,* , Andreas C. Lazaris 1, Konstantinos Arsenoglou 2, Maria Emmanouil 2, Olympia Kyriakides 3, Nikolaos Kavantzas 1 and Irene Panderi 4,* 1 Laboratory of Pathological Anatomy, Department of Clinical and Laboratory Medicine, School of Medicine, National and Kapodistrian University of Athens, 75, Mikras Asias Avenue, Goudi, 11527 Athens, Greece; [email protected] (A.C.L.); [email protected] (N.K.) 2 Veterinary Services, Ministry of Agriculture, Rural Development and Environment, 1417 Nicosia, Cyprus; [email protected] (K.A.); [email protected] (M.E.) 3 Archbishop Makarios III Hospital, 2012 Nicosia, Cyprus; [email protected] 4 Laboratory of Pharmaceutical Analysis, Panepistimiopolis, Division of Pharmaceutical Chemistry, Department of Pharmacy, National and Kapodistrian University of Athens, Zografou, 15771 Athens, Greece * Correspondence: [email protected] (D.K.); [email protected] (I.P.); Tel.: +30-210-727-4820 (I.P.) Received: 1 September 2020; Accepted: 13 October 2020; Published: 16 October 2020 Abstract: In recent years, huge amounts of antibiotics have been administered to farm animals, and as a result, residues of these antibiotics can accumulate in livestock products and, once consumed, may be transmitted to humans. Farm animals’ antibiotic treatment may therefore present a risk for consumers health, especially for children and adolescents. In children, the immune system is not fully developed, and thus, they are more susceptible than adults to resistant bacteria. A dietary exposure assessment was conducted on veterinary antibiotics found in raw pork meat among children and adolescents in Cyprus, since pork is the most consumed red meat in Cypriot population. -

The Ocean As a Global Reservoir of Antibiotic Resistance Genes
The Ocean as a Global Reservoir of Antibiotic Resistance Genes Stephen M. Hatosy,a Adam C. Martinya,b Department of Ecology and Evolutionary Biology, University of California, Irvine, California, USAa; Department of Earth System Science, University of California, Irvine, California, USAb Recent studies of natural environments have revealed vast genetic reservoirs of antibiotic resistance (AR) genes. Soil bacteria and Downloaded from human pathogens share AR genes, and AR genes have been discovered in a variety of habitats. However, there is little knowledge about the presence and diversity of AR genes in marine environments and which organisms host AR genes. To address this, we identified the diversity of genes conferring resistance to ampicillin, tetracycline, nitrofurantoin, and sulfadimethoxine in diverse marine environments using functional metagenomics (the cloning and screening of random DNA fragments). Marine environ- ments were host to a diversity of AR-conferring genes. Antibiotic-resistant clones were found at all sites, with 28% of the genes identified as known AR genes (encoding beta-lactamases, bicyclomycin resistance pumps, etc.). However, the majority of AR genes were not previously classified as such but had products similar to proteins such as transport pumps, oxidoreductases, and hydrolases. Furthermore, 44% of the genes conferring antibiotic resistance were found in abundant marine taxa (e.g., Pelagibac- http://aem.asm.org/ ter, Prochlorococcus, and Vibrio). Therefore, we uncovered a previously unknown diversity of genes that conferred an AR pheno- type among marine environments, which makes the ocean a global reservoir of both clinically relevant and potentially novel AR genes. he spread of antibiotic resistance (AR) is critically important sity of marine AR genes, and (iii) are these genes harbored by Tto human health. -

Tetracycline and Sulfonamide Antibiotics in Soils: Presence, Fate and Environmental Risks
processes Review Tetracycline and Sulfonamide Antibiotics in Soils: Presence, Fate and Environmental Risks Manuel Conde-Cid 1, Avelino Núñez-Delgado 2 , María José Fernández-Sanjurjo 2 , Esperanza Álvarez-Rodríguez 2, David Fernández-Calviño 1,* and Manuel Arias-Estévez 1 1 Soil Science and Agricultural Chemistry, Faculty Sciences, University Vigo, 32004 Ourense, Spain; [email protected] (M.C.-C.); [email protected] (M.A.-E.) 2 Department Soil Science and Agricultural Chemistry, Engineering Polytechnic School, University Santiago de Compostela, 27002 Lugo, Spain; [email protected] (A.N.-D.); [email protected] (M.J.F.-S.); [email protected] (E.Á.-R.) * Correspondence: [email protected] Received: 30 October 2020; Accepted: 13 November 2020; Published: 17 November 2020 Abstract: Veterinary antibiotics are widely used worldwide to treat and prevent infectious diseases, as well as (in countries where allowed) to promote growth and improve feeding efficiency of food-producing animals in livestock activities. Among the different antibiotic classes, tetracyclines and sulfonamides are two of the most used for veterinary proposals. Due to the fact that these compounds are poorly absorbed in the gut of animals, a significant proportion (up to ~90%) of them are excreted unchanged, thus reaching the environment mainly through the application of manures and slurries as fertilizers in agricultural fields. Once in the soil, antibiotics are subjected to a series of physicochemical and biological processes, which depend both on the antibiotic nature and soil characteristics. Adsorption/desorption to soil particles and degradation are the main processes that will affect the persistence, bioavailability, and environmental fate of these pollutants, thus determining their potential impacts and risks on human and ecological health. -

Sulfadimethoxine Degradation Kinetics in Manure As Affected by Initial Concentration, Moisture, and Temperature
Published online October 27, 2006 Sulfadimethoxine Degradation Kinetics in Manure as Affected by Initial Concentration, Moisture, and Temperature Q.-Q. Wang, S. A. Bradford, W. Zheng, and S. R. Yates* ABSTRACT However in Europe, a new trend has developed with in- Sulfadimethoxine is a widely used sulfonamide veterinary antibiotic creased therapeutic use of antibiotics (Alder et al., 2001). and could be a source of agricultural contamination. Therefore, infor- After the application to animals, antibiotics will even- mation is needed about its degradation kinetics in manure under aer- tually enter the environment. In fish farming, antibiotics obic conditions. Based on the analysis of first-order kinetics and the are given as feed additives, resulting in a direct release assumption that sulfadimethoxine availability for degradation in ma- of antibiotics into the aquatic environment (Thurman nure could be limiting, a new kinetic model was developed and was et al., 2002). It was estimated that |70 to 80% of drugs found to fit the degradation kinetics well. The degradation rate in administered to fish enters the environment and anti- sterile manure was found to be much lower than in nonsterile manure, biotic residues with significant antibacterial activity were indicating that biodegradation was significant. In biologically active found in the sediment of fish hatcheries (Samuelsen et al., manure, the degradation rate constant decreased with increasing ini- tial concentration of sulfadimethoxine, implying that the activity of 1992). However, the major route through which veteri- the degrading microorganisms was inhibited. Increasing moisture or nary antibiotics enter the environment is the excretion temperature was found to increase sulfadimethoxine degradation in of feces and urine from medicated animals in livestock manure. -

2019-6654 Final Report of an Audit Carried out In
Ref. Ares(2019)7826731 - 19/12/2019 EUROPEAN COMMISSION DIRECTORATE-GENERAL FOR HEALTH AND FOOD SAFETY Health and food audits and analysis DG(SANTE) 2019-6654 FINAL REPORT OF AN AUDIT CARRIED OUT IN BELARUS FROM 13 TO 24 MAY 2019 IN ORDER TO EVALUATE THE CONTROL OF RESIDUES AND CONTAMINANTS IN LIVE ANIMALS AND ANIMAL PRODUCTS INCLUDING CONTROLS ON VETERINARY MEDICINAL PRODUCTS Executive Summary This report describes the outcome of an audit carried out in Belarus from 13 to 24 May 2019 as part of the European Commission’s Directorate-General for Health and Food Safety planned work programme. The objective of the audit was to evaluate the effectiveness of official controls on residues and contaminants in live animals and animal products eligible for export to the European Union (EU). The audit assessed the implementation of the residue monitoring plan and also covered the authorisation, distribution and use of veterinary medicinal products, given that these areas have an impact on the monitoring of residues. Attention was also paid to examining the implementation of corrective actions indicated in response to specific recommendations made in the report of the previous residues audit to Belarus. The planning of the residue monitoring largely follows the principles of Directive 96/23/EC and covers for the most part an appropriate range of substances. The plan is nevertheless weakened by the fact that action levels for several substances across all commodities (including those for which listing has been requested) are not aligned with EU maximum residue limits, thus the plan would not be sufficient to demonstrate that commodities eligible for export to the EU would comply with such limits where they are lower that national limits. -

Use of Antibiotics in Ornamental Fish Aquaculture1 Roy P
Cir 84 Use of Antibiotics in Ornamental Fish Aquaculture1 Roy P. E. Yanong2 Introduction based on their response to a protocol called gram staining. Gram-positive bacteria stain blue, and gram-negative Antibiotics are very useful additions to any fish-health bacteria stain pink. They stain differently because each manager’s toolbox, but they are only tools and not ‘magic group has a different type of outer structure known as the bullets.’ The ability of antibiotics to help eliminate a fish cell wall. This difference is important for the producer disease depends on a number of factors: 1) Does the and aquaculturist because some antibiotics work better problem actually have a bacterial component? 2) Are the against gram-positive bacteria and others work better bacteria involved sensitive to the antibiotic chosen? 3) Are against gram-negative bacteria. Most bacteria that infect the proper dosage and treatment intervals being used? 4) fish are gram-negative, including Aeromonas hydrophila, Have other contributing stresses been removed or reduced? Aeromonas salmonicida, Flavobacterium columnare (which causes columnaris), Vibrio, and Pseudomonas species. (See Antibiotics, in and of themselves, do not cure a fish. Antibi- UF/IFAS Fact Sheets FA-14 Aeromonas Infections, FA-31 otics merely control the population growth of bacteria in a Vibrio Infections of Fish and FA-11 Columnaris disease). fish long enough for its immune system to eliminate them. The major group of gram-positive bacteria that cause Before antibiotics are even considered, sources of stress disease in fish are Streptococcus. (See UF/IFAS Circular 57 such as poor water quality (including drastic temperature Streptococcal Infections in Fish.) change), nutrition, genetics, and handling or transport must A third group, the acid-fast bacteria, which includes be removed or reduced. -

Sulfonamides and Sulfonamide Combinations*
Sulfonamides and Sulfonamide Combinations* Overview Due to low cost and relative efficacy against many common bacterial infections, sulfonamides and sulfonamide combinations with diaminopyrimidines are some of the most common antibacterial agents utilized in veterinary medicine. The sulfonamides are derived from sulfanilamide. These chemicals are structural analogues of ρ-aminobenzoic acid (PABA). All sulfonamides are characterized by the same chemical nucleus. Functional groups are added to the amino group or substitutions made on the amino group to facilitate varying chemical, physical and pharmacologic properties and antibacterial spectra. Most sulfonamides are too alkaline for routine parenteral use. Therefore the drug is most commonly administered orally except in life threatening systemic infections. However, sulfonamide preparations can be administered orally, intramuscularly, intravenously, intraperitoneally, intrauterally and topically. Sulfonamides are effective against Gram-positive and Gram-negative bacteria. Some protozoa, such as coccidians, Toxoplasma species and plasmodia, are generally sensitive. Chlamydia, Nocardia and Actinomyces species are also sensitive. Veterinary diseases commonly treated by sulfonamides are actinobacillosis, coccidioidosis, mastitis, metritis, colibacillosis, pododermatitis, polyarthritis, respiratory infections and toxo- plasmosis. Strains of rickettsiae, Pseudomonas, Klebsiella, Proteus, Clostridium and Leptospira species are often highly resistant. Sulfonamides are bacteriostatic antimicrobials -

On the Extraction of Antibiotics from Shrimps Prior to Chromatographic Analysis
separations Review On the Extraction of Antibiotics from Shrimps Prior to Chromatographic Analysis Victoria Samanidou *, Dimitrios Bitas, Stamatia Charitonos and Ioannis Papadoyannis Laboratory of Analytical Chemistry, University of Thessaloniki, GR 54124 Thessaloniki, Greece; [email protected] (D.B.); [email protected] (S.C.); [email protected] (I.P.) * Correspondence: [email protected]; Tel.: +30-2310-997698; Fax: +30-2310-997719 Academic Editor: Frank L. Dorman Received: 2 February 2016; Accepted: 26 February 2016; Published: 4 March 2016 Abstract: The widespread use of antibiotics in veterinary practice and aquaculture has led to the increase of antimicrobial resistance in food-borne pathogens that may be transferred to humans. Global concern is reflected in the regulations from different agencies that have set maximum permitted residue limits on antibiotics in different food matrices of animal origin. Sensitive and selective methods are required to monitor residue levels in aquaculture species for routine regulatory analysis. Since sample preparation is the most important step, several extraction methods have been developed. In this review, we aim to summarize the trends in extraction of several antibiotics classes from shrimps and give a comparison of performance characteristics in the different approaches. Keywords: sample preparation; extraction; aquaculture; shrimps; chromatography; antibiotics 1. Introduction According to FAO (CWP Handbook of Fishery Statistical Standards, Section J: AQUACULTURE), “aquaculture is the farming of aquatic organisms: fish, mollusks, crustaceans, aquatic plants, crocodiles, alligators, turtles, and amphibians. Farming implies some form of intervention in the rearing process to enhance production, such as regular stocking, feeding, protection from predators, etc.”[1]. Since 1960, aquaculture practice and production has increased as a result of the improved conditions in the aquaculture facilities. -
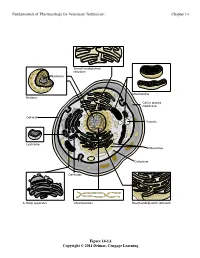
Fundamentals of Pharmacology for Veterinary Technicians Chapter 14
Fundamentals of Pharmacology for Veterinary Technicians Chapter 14 6PRRWKHQGRSODVPLF UHWLFXOXP 1XFOHROXV 0LWRFKRQGULD 1XFOHXV &HOORUSODVPD PHPEUDQH &HOOZDOO 9DFXROH /\VRVRPH 5LERVRPHV &\WRSODVP &HQWULROHV $*ROJLDSSDUDWXV &KURPRVRPHV 5RXJKHQGRSODVPLFUHWLFXOXP Figure 14-1A Copyright © 2011 Delmar, Cengage Learning Fundamentals of Pharmacology for Veterinary Technicians Chapter 14 1XFOHRLG '1$ 3LOXV 5LERVRPHV )ODJHOOXP &DSVXOH &HOOZDOO &HOOPHPEUDQH 3ODVPLG '1$ &\WRSODVP % Figure 14-1B Copyright © 2011 Delmar, Cengage Learning Fundamentals of Pharmacology for Veterinary Technicians Chapter 14 6WHS 3URFHGXUH 5HVXOW 3ULPDU\VWDLQ$SSO\FU\VWDO $OOEDFWHULDVWDLQSXUSOH YLROHWVWDLQ SXUSOH 5LQVHVOLGH 0RUGDQW$SSO\*UDP VLRGLQH $OOEDFWHULDUHPDLQSXUSOH 5LQVHVOLGH 'HFRORUL]H$SSO\DFLGDOFRKRORUDFHWRQH 3XUSOHVWDLQLVUHPRYHGIURP JUDPQHJDWLYHFHOOV 5LQVHVOLGH &RXQWHUVWDLQ$SSO\VDIUDQLQ *UDPQHJDWLYHFHOOVDSSHDU VWDLQ UHG SLQNUHGJUDPSRVLWLYHFHOOV DSSHDUSXUSOH 5LQVHVOLGH Figure 14-2 Copyright © 2011 Delmar, Cengage Learning Fundamentals of Pharmacology for Veterinary Technicians Chapter 14 Figure 14-3 Copyright © 2011 Delmar, Cengage Learning Fundamentals of Pharmacology for Veterinary Technicians Chapter 14 +JRIDQWLELRWLFSHUP/RIEURWK Figure 14-4 Copyright © 2011 Delmar, Cengage Learning Fundamentals of Pharmacology for Veterinary Technicians Chapter 14 1XFOHLFDFLG V\QWKHVLVDOWHUDWLRQ 1XFOHRLG '1$ 3URWHLQV\QWKHVLV LQKLELWLRQ 3LOXV 5LERVRPHV %ORFNLQJWKHXVH RIQHHGHGQXWULHQWV )ODJHOOXP &DSVXOH &HOOZDOO 3ODVPLG '1$ &\WRSODVP &HOOZDOO GLVUXSWLRQ &HOOPHPEUDQH &HOOPHPEUDQH -

Alphabetical Listing of ATC Drugs & Codes
Alphabetical Listing of ATC drugs & codes. Introduction This file is an alphabetical listing of ATC codes as supplied to us in November 1999. It is supplied free as a service to those who care about good medicine use by mSupply support. To get an overview of the ATC system, use the “ATC categories.pdf” document also alvailable from www.msupply.org.nz Thanks to the WHO collaborating centre for Drug Statistics & Methodology, Norway, for supplying the raw data. I have intentionally supplied these files as PDFs so that they are not quite so easily manipulated and redistributed. I am told there is no copyright on the files, but it still seems polite to ask before using other people’s work, so please contact <[email protected]> for permission before asking us for text files. mSupply support also distributes mSupply software for inventory control, which has an inbuilt system for reporting on medicine usage using the ATC system You can download a full working version from www.msupply.org.nz Craig Drown, mSupply Support <[email protected]> April 2000 A (2-benzhydryloxyethyl)diethyl-methylammonium iodide A03AB16 0.3 g O 2-(4-chlorphenoxy)-ethanol D01AE06 4-dimethylaminophenol V03AB27 Abciximab B01AC13 25 mg P Absorbable gelatin sponge B02BC01 Acadesine C01EB13 Acamprosate V03AA03 2 g O Acarbose A10BF01 0.3 g O Acebutolol C07AB04 0.4 g O,P Acebutolol and thiazides C07BB04 Aceclidine S01EB08 Aceclidine, combinations S01EB58 Aceclofenac M01AB16 0.2 g O Acefylline piperazine R03DA09 Acemetacin M01AB11 Acenocoumarol B01AA07 5 mg O Acepromazine N05AA04 -

Federal Register / Vol. 60, No. 80 / Wednesday, April 26, 1995 / Notices DIX to the HTSUS—Continued
20558 Federal Register / Vol. 60, No. 80 / Wednesday, April 26, 1995 / Notices DEPARMENT OF THE TREASURY Services, U.S. Customs Service, 1301 TABLE 1.ÐPHARMACEUTICAL APPEN- Constitution Avenue NW, Washington, DIX TO THE HTSUSÐContinued Customs Service D.C. 20229 at (202) 927±1060. CAS No. Pharmaceutical [T.D. 95±33] Dated: April 14, 1995. 52±78±8 ..................... NORETHANDROLONE. A. W. Tennant, 52±86±8 ..................... HALOPERIDOL. Pharmaceutical Tables 1 and 3 of the Director, Office of Laboratories and Scientific 52±88±0 ..................... ATROPINE METHONITRATE. HTSUS 52±90±4 ..................... CYSTEINE. Services. 53±03±2 ..................... PREDNISONE. 53±06±5 ..................... CORTISONE. AGENCY: Customs Service, Department TABLE 1.ÐPHARMACEUTICAL 53±10±1 ..................... HYDROXYDIONE SODIUM SUCCI- of the Treasury. NATE. APPENDIX TO THE HTSUS 53±16±7 ..................... ESTRONE. ACTION: Listing of the products found in 53±18±9 ..................... BIETASERPINE. Table 1 and Table 3 of the CAS No. Pharmaceutical 53±19±0 ..................... MITOTANE. 53±31±6 ..................... MEDIBAZINE. Pharmaceutical Appendix to the N/A ............................. ACTAGARDIN. 53±33±8 ..................... PARAMETHASONE. Harmonized Tariff Schedule of the N/A ............................. ARDACIN. 53±34±9 ..................... FLUPREDNISOLONE. N/A ............................. BICIROMAB. 53±39±4 ..................... OXANDROLONE. United States of America in Chemical N/A ............................. CELUCLORAL. 53±43±0 -

Concept Paper
Concept Paper: Potential Approach for Ranking of Antimicrobial Drugs According to Their Importance in Human Medicine: A Risk Management Tool for Antimicrobial New Animal Drugs Introduction This concept paper discusses a potential approach to considering the human medical importance of antimicrobial drugs when assessing and managing antimicrobial resistance risks associated with the use of antimicrobial drugs in animals. In 2003, FDA established a list ranking antimicrobial drugs according to their relative importance in human medicine primarily for the purpose of supporting a qualitative risk assessment process outlined in agency guidance. It was envisioned by the Agency at the time the current medical importance rankings list was published that it would periodically reassess the rankings to align with contemporary science and current human clinical practices. To that end, this paper describes a potential revised process for ranking antimicrobial drugs according to their relative importance in human medicine, potential revised criteria to determine the medical importance rankings, and the list of antimicrobial drug medical importance rankings that would result if those criteria were to be used. Disclaimer: This concept paper is for discussion purposes only. The intent of this concept paper is to obtain public comment and early input on a potential approach to consider the human medical importance of antimicrobial drugs when assessing and managing antimicrobial resistance risks associated with the use of antimicrobial drugs in animals. This concept paper does not contain recommendations and does not constitute draft or final guidance by the Food and Drug Administration. It should not be used for any purpose other than to facilitate public comment.