Linux for Biologists
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

110% Gaming 220 Triathlon Magazine 3D World Adviser
110% Gaming 220 Triathlon Magazine 3D World Adviser Evolution Air Gunner Airgun World Android Advisor Angling Times (UK) Argyllshire Advertiser Asian Art Newspaper Auto Car (UK) Auto Express Aviation Classics BBC Good Food BBC History Magazine BBC Wildlife Magazine BIKE (UK) Belfast Telegraph Berkshire Life Bikes Etc Bird Watching (UK) Blackpool Gazette Bloomberg Businessweek (Europe) Buckinghamshire Life Business Traveller CAR (UK) Campbeltown Courier Canal Boat Car Mechanics (UK) Cardmaking and Papercraft Cheshire Life China Daily European Weekly Classic Bike (UK) Classic Car Weekly (UK) Classic Cars (UK) Classic Dirtbike Classic Ford Classic Motorcycle Mechanics Classic Racer Classic Trial Classics Monthly Closer (UK) Comic Heroes Commando Commando Commando Commando Computer Active (UK) Computer Arts Computer Arts Collection Computer Music Computer Shopper Cornwall Life Corporate Adviser Cotswold Life Country Smallholding Country Walking Magazine (UK) Countryfile Magazine Craftseller Crime Scene Cross Stitch Card Shop Cross Stitch Collection Cross Stitch Crazy Cross Stitch Gold Cross Stitcher Custom PC Cycling Plus Cyclist Daily Express Daily Mail Daily Star Daily Star Sunday Dennis the Menace & Gnasher's Epic Magazine Derbyshire Life Devon Life Digital Camera World Digital Photo (UK) Digital SLR Photography Diva (UK) Doctor Who Adventures Dorset EADT Suffolk EDGE EDP Norfolk Easy Cook Edinburgh Evening News Education in Brazil Empire (UK) Employee -

Pressreader Magazine Titles
PRESSREADER: UK MAGAZINE TITLES www.edinburgh.gov.uk/pressreader Computers & Technology Sport & Fitness Arts & Crafts Motoring Android Advisor 220 Triathlon Magazine Amateur Photographer Autocar 110% Gaming Athletics Weekly Cardmaking & Papercraft Auto Express 3D World Bike Cross Stitch Crazy Autosport Computer Active Bikes etc Cross Stitch Gold BBC Top Gear Magazine Computer Arts Bow International Cross Stitcher Car Computer Music Boxing News Digital Camera World Car Mechanics Computer Shopper Carve Digital SLR Photography Classic & Sports Car Custom PC Classic Dirt Bike Digital Photographer Classic Bike Edge Classic Trial Love Knitting for Baby Classic Car weekly iCreate Cycling Plus Love Patchwork & Quilting Classic Cars Imagine FX Cycling Weekly Mollie Makes Classic Ford iPad & Phone User Cyclist N-Photo Classics Monthly Linux Format Four Four Two Papercraft Inspirations Classic Trial Mac Format Golf Monthly Photo Plus Classic Motorcycle Mechanics Mac Life Golf World Practical Photography Classic Racer Macworld Health & Fitness Simply Crochet Evo Maximum PC Horse & Hound Simply Knitting F1 Racing Net Magazine Late Tackle Football Magazine Simply Sewing Fast Bikes PC Advisor Match of the Day The Knitter Fast Car PC Gamer Men’s Health The Simple Things Fast Ford PC Pro Motorcycle Sport & Leisure Today’s Quilter Japanese Performance PlayStation Official Magazine Motor Sport News Wallpaper Land Rover Monthly Retro Gamer Mountain Biking UK World of Cross Stitching MCN Stuff ProCycling Mini Magazine T3 Rugby World More Bikes Tech Advisor -
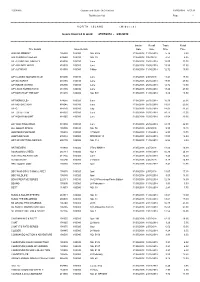
Distribution List Page - 1
R56891A Gordon and Gotch (NZ) Limited 29/05/2018 8:57:12 Distribution List Page - 1 =========================================================================================================================================================== N O R T H I S L A N D ( M i d w e e k ) Issues invoiced in week 27/05/2018 - 2/06/2018 =========================================================================================================================================================== Invoice Recall Trade Retail Title Details Issue Details Date Date Price Price 2000 AD WEEKLY 105030 100080 NO. 2075 31/05/2018 11/06/2018 5.74 8.80 500 SUDOKU PUZZLES 239580 100010 NO. 39 31/05/2018 9/07/2018 4.53 6.95 A/F AEROPLANE MNTHLY 818005 100010 June 31/05/2018 25/06/2018 15.00 23.00 A/F AIRLINER WRLD 818020 100020 June 31/05/2018 25/06/2018 15.59 23.90 A/F AUTOCAR 818030 100080 9 May 31/05/2018 11/06/2018 12.72 19.50 A/F CLASSIC MOTORCYCLE 915600 100010 June 31/05/2018 2/07/2018 11.41 17.50 A/F GUITARIST 818155 100010 June 31/05/2018 25/06/2018 17.93 27.50 A/F INSIDE UNITED 896390 100010 June 31/05/2018 25/06/2018 12.72 19.50 A/F LINUX FORMAT DVD 818185 100010 June 31/05/2018 25/06/2018 18.20 27.90 A/F MATCH OF THE DAY 818215 100080 NO. 504 31/05/2018 11/06/2018 8.80 13.50 A/F MONOCLE> 874655 100020 June 31/05/2018 25/06/2018 16.30 25.00 A/F MOTORSPORT 818245 100010 June 31/05/2018 25/06/2018 13.63 20.90 A/F Q 818270 100010 July 31/05/2018 25/06/2018 14.02 21.50 A/F TOTAL FILM 818335 100010 June 31/05/2018 25/06/2018 10.11 15.50 A/F WOMAN&HOME 818365 100010 June 31/05/2018 25/06/2018 13.04 20.00 A/F YACHTING WRLD 818380 100010 June 31/05/2018 25/06/2018 14.35 22.00 ALL ABOUT SPACE 105505 100010 NO. -

Photini Documentation Release 2017.6.0.Dev819
Photini Documentation Release 2017.6.0.dev819 Jim Easterbrook Jun 02, 2017 Contents 1 Introduction 3 1.1 Features..................................................4 1.2 Dependencies...............................................5 1.3 Getting help...............................................6 1.4 Internationalisation............................................6 1.5 Licence..................................................6 2 Installation 9 2.1 All-in-one installer (Windows)......................................9 2.2 Package manager (some Linux distributions).............................. 10 2.3 Dependencies package (some Linux distributions)........................... 10 2.4 Piecemeal installation.......................................... 11 2.5 Essential dependencies.......................................... 11 2.6 Installing Photini............................................. 12 2.7 Running Photini............................................. 14 2.8 Photini documentation.......................................... 14 3 User manual 17 3.1 Image selector.............................................. 17 3.2 Descriptive metadata........................................... 23 3.3 Technical metadata............................................ 29 3.4 Geotagging................................................ 39 3.5 Flickr uploader.............................................. 53 3.6 Google Photos / Picasa uploader..................................... 60 3.7 Facebook uploader............................................ 66 3.8 Image importer............................................. -

Linux at The
Linux at the BBC Linux at the BBC What do Strictly Come Dancing, the shipping forecast and open source have in common? They’re all big at the BBC, as Alex Singleton finds out. he British Broadcasting Corporation is the biggest broadcaster in the world. It spends more than £4bn a year and employs 28,000 people. It transmits eight UK television channels, six overseas T channels, countless radio stations in over 200 countries, and has a website with over two million pages. As you’d expect, the BBC’s technical requirements are rather different from those of the average business IT user. Its support contract with Siemens is worth nearly £200m a year, for example, and a technical fault can mean millions of people looking at a blank screen, either on their TV or their computer. The BBC’s research and development team is based at Kingswood Warren, a large country mansion in Surrey. For the staff at Kingswood, Linux is the operating system of choice. They prefer it as a development platform and they find that demanding software runs better on it than it does on Windows. Here Linux-based applications are being developed that push the operating filming with several cameras can easily result in 35 hours of recorded material. system’s use out of its traditional niches and into the heart of the BBC’s work. If recording takes a week, programme-makers can wait days for the tapes to be inputted into an editing system. Ingex eliminates that process by feeding Better for broadcasting the camera output directly into a Linux-based computer with a gigantic hard The BBC’s empire stretches computer equipment to its limits: if a studio is disk. -

(OR LESS!) Food & Cooking English One-Off (Inside) Interior Design
Publication Magazine Genre Frequency Language $10 DINNERS (OR LESS!) Food & Cooking English One-Off (inside) interior design review Art & Photo English Bimonthly . -

Build Your Own Distro Tired of the Run-Of-The-Mill Stuff on Distrowatch? Why Not Heed Mayank Sharma and Create the Perfect Operating System for You?
Build your own distro Tired of the run-of-the-mill stuff on Distrowatch? Why not heed Mayank Sharma and create the perfect operating system for you? few issues ago, [LXF171, 50 making it your own – by removing apps and Now traditional wisdom says that creating Distros Tested], we looked at drivers that you don’t need and adding the your own Linux system is a rather difficult the best Linux distributions for ones you do. You’ll also probably change the thing to do and shouldn’t be attempted by A all kinds of users. There were factory-fitted artwork that says more about anyone other than Linux veterans. distros that were designed with ease of use the distro vendor than you. We begin the feature with graphical point- in mind, some focused on productivity, Sure that’s one way to go about it. and-click tools. Yes, you read that right. All it while others catered to specialised use You tweak and customise the distro to suit takes is a couple of clicks to craft your very cases, such as security and privacy your requirements. But wouldn’t it be really own flavour of Linux that you can pass to conscious users. But we can guarantee great if you just create your very own, custom friends and family. We’ve got tools that’ll help that there wasn’t a distro designed Linux distribution? you create and distribute customised spins specifically and entirely for you! based on Ubuntu, Fedora and While most Linux users make do OpenSUSE – the three mainstream with one of the mainstream distros out “We’ve got tools that’ll Linux distributions that house there, there’s always something or the help you create and thousands of open source software in other that’s missing. -

Future Publishing Statement for IPSO – April 2016
Future Publishing Statement for IPSO – April 2016 About Future Future plc is an international publishing and media group, and a leading digital company. Celebrating over 30 years in business, Future was founded in 1985 with one magazine – we now create over 200 print publications, apps, websites and events through operations in the UK, US and Australia. The company employs approximately 500 employees. Future’s leadership structure is outlined in Appendix C (the structure has changed slightly since our last statement in December 2015). Our portfolio covers consumer technology, games/entertainment, music, creative/design and photography. 48 million users globally access Future’s digital sites each month, we have over 200,000 digital subscriptions worldwide, and a combined social media audience of 20+ million followers (a list of our titles/products can be found under Appendix A). For the purpose of this statement, Future’s ‘responsible person’ is Nial Ferguson, Content Director, Media. Future’s editorial standards Through our expertise in five portfolios, Future produces engaging, informative and entertaining content to a high standard. The business is driven by a core strategy - ‘Content that Connects’ - that has been in place since 2014 (see Appendix B). This puts content at the heart of what we do, and is an approach we reiterate through staff communication. In creating content that connects, Future takes all reasonable and appropriate steps to verify what we publish. Such steps include double sourcing where necessary, and rigorous scrutiny of information and sources to ensure the accuracy of the articles we publish. Editorial process for contentious issues involves second reading by editorial. -

Volume 50 March, 2011
Volume 50 March, 2011 e17: Running Ecomorph e17: Configure The Everything Module To Do Everything e17: Settings Panel, Part 3 Free At Last! LibreOffice 3.3 Released Synaptic & The Repositories Video Part 1: Time Shifting TV Programs Video Part 2: Editing Your Recorded Video Avidemux Under PCLinuxOS Alternate OS: Icaros, Part 1 New Forum Launched: PCLinuxOS Kids WindowMaker on PCLinuxOS: The Basics Ladies Of PCLinuxOS: ms_meme PCLinuxOS & Linux In Greece Using Scribus, Part 3: Text! Text! Text! And much more inside! TTaabbllee OOff CCoonntteennttss 3 Welcome From The Chief Editor 5 e17: Configure Everything To Do Everything 8 Scribus, Part 3: Text, Text, Text 10 PCLinuxOS & Linux In Greece The PCLinuxOS name, logo and colors are the trademark of 11 Screenshot Showcase Texstar. 12 e17: Settings Panel, Part 3 The NEW PCLinuxOS Magazine is a monthly online 14 Ladies Of PCLinuxOS: ms_meme publication containing PCLinuxOSrelated materials. It is published primarily for members of the PCLinuxOS 15 Double Take & Mark's Quick Gimp Tip community. The Magazine staff is comprised of volunteers 16 e17: Running Ecomorph from the PCLinuxOS community. 19 Screenshot Showcase Visit us online at http://www.pclosmag.com 20 Forum Foibles: Roman Holiday This release was made possible by the following volunteers: 21 Video Part 1: Time Shifting TV Programs Chief Editor: Paul Arnote (parnote) Assistant Editors: Andrew Strick (Stricktoo), Meemaw 24 Screenshot Showcase Consultants: Archie Arevalo, Tim Robinson 25 New Forum Launched: PCLinuxOS Kids Artwork: Sproggy, Timeth, Mark Szorady Magazine Layout: parnote, Meemaw, ms_meme, Stricktoo 26 Video Part 2: Editing Your Recorded Video HTML Layout: Galen Seaman 29 Screenshot Showcase Staff: 30 Free At Last! LibreOffice 3.3 Released Neal Brooks ms_meme Galen Seaman Mark Szorady 33 Screenshot Showcase Patrick Horneker Darrel Johnston Guy Taylor Meemaw 34 Alternate OS: Icaros, Part 1 Andrew Huff Gary L. -

Rbdigital Magazines Titles by Genre June 2019
RBdigital Magazines Titles by Genre June 2019 Title Country Genre Architectural Digest United States Architecture Architecture Australia Australia Architecture Interior New Zealand Architecture Amateur Photographer United Kingdom Art & Photo Aperture United States Art & Photo Art New Zealand New Zealand Art & Photo ArtAsiaPacific New Hong Kong Art & Photo Artist Profile Australia Art & Photo Artists & Illustrators United Kingdom Art & Photo Artist's Magazine, The United Kingdom Art & Photo Australian Photography + Digital Australia Art & Photo Camera Australia Art & Photo Digital Camera World United Kingdom Art & Photo Digital Photo United States Art & Photo Digital Photographer United Kingdom Art & Photo D-Photo New Zealand Art & Photo Dumbo Feather Australia Art & Photo Harper's Magazine United States Art & Photo Magazine Antiques, The United States Art & Photo Mark Magazine Netherlands Art & Photo Wallpaper United Kingdom Art & Photo RBdigital Magazines Titles by Genre June 2019 Title Country Genre Australian Muscle Car Australia Automotive Auto Express United Kingdom Automotive Autocar United Kingdom Automotive Automobile United States Automotive BBC Top Gear Magazine United Kingdom Automotive CAR United Kingdom Automotive Classic & Sports Car United Kingdom Automotive Classic Cars United Kingdom Automotive Classic Driver New Zealand Automotive Classic Trucks United States Automotive Classics Monthly United Kingdom Automotive Evo United Kingdom Automotive F1 Racing UK United Kingdom Automotive Hot Rod United States Automotive Motor -

Using Zcluster Georgia Advanced Computing
Introduction to Linux Basics Part-I Georgia Advanced Computing Resource Center University of Georgia Zhuofei Hou, HPC Trainer [email protected] Outline • What is GACRC? • What is Linux? • Linux Command, Shell and Filesystem Concepts • Linux Common Commands What is GACRC? Who Are We? Georgia Advanced Computing Resource Center Collaboration between the Office of Vice President for Research (OVPR) and the Office of the Vice President for Information Technology (OVPIT) Guided by a faculty advisory committee (GACRC-AC) Why Are We Here? To provide computing hardware and network infrastructure in support of high- performance computing (HPC) at UGA Where Are We? http://gacrc.uga.edu (Web) http://wiki.gacrc.uga.edu (Wiki) https://wiki.gacrc.uga.edu/wiki/Getting_Help (Support) https://blog.gacrc.uga.edu (Blog) http://forums.gacrc.uga.edu (Forums) What is Linux? • What is Operating System (OS)? • What is Linux OS? • Brief History of Linux OS • Why Linux OS? What is Linux – Operating System Operating System (OS) : Program initially loaded at booting time, to manage all the other application programs on a computer Software interface between computer hardware and its human user Needed for ALL computers to be operated Needed to run software and control hardware Popular OSes: What is Linux – Linux OS Linux OS is a full-fledged OS with 4 major parts: I. Kernel: Low-level OS, handling files, disks, RAM, networking, etc. II. Supplied Programs: Web browsing, Audio, Video, DVD burning…… III. The Shell: A command-line user interface for a user to type and execute commands: Bourne Shell (sh) Korn Shell (ksh) UNIX standard shells C Shell (csh) Bourne-Again Shell (bash) Linux default shell IV. -

Raspberry Pi Add-Ons Ben Everard Investigates the Extras on Offer for the Device That Has Turned Us All Into Tinkerers
Raspberry Pi add-ons Ben Everard investigates the extras on offer for the device that has turned us all into tinkerers. he Raspberry Pi is probably the most understand how to use it, though. This was a these haven’t been developed by large successful British computing product particular problem when the Raspberry Pi first companies, but by hobbyists who saw a need Tin a decade, but it’s also one of the came out: there were few tutorials explaining and filled it. It’s been great to see just how most misunderstood. Too many people think of how to use it, and if you wanted to use the quickly new and innovative devices have come the Pi as just a cheap desktop, but by the time GPIO you had to build any add-ons for yourself. on to the market. you’ve bought a monitor, keyboard, mouse and Fortunately, times have changed quite rapidly, Here, we’re going to look at three of our SD card, you’ll have spent almost as much as and a whole ecosystem of components for the favourite add-ons, so if you’ve got a Raspberry you would buying a cheap laptop – and it’s a Pi has sprung up. Pi acting as a paperweight, blow off the dust whole lot less powerful. Every day, it seems, we hear news of some and put it to a more productive use; if your Pi The real innovation of the Pi, then, isn’t its new device that connects to the Pi to enable project is already complete, well, you know it cost, but its form factor.