The Solution Structure of the Guanine Nucleotide Exchange Domain Of
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Initiation Factor Eif5b Catalyzes Second GTP-Dependent Step in Eukaryotic Translation Initiation
Initiation factor eIF5B catalyzes second GTP-dependent step in eukaryotic translation initiation Joon H. Lee*†, Tatyana V. Pestova†‡§, Byung-Sik Shin*, Chune Cao*, Sang K. Choi*, and Thomas E. Dever*¶ *Laboratory of Gene Regulation and Development, National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, MD 20892-2716; ‡Department of Microbiology and Immunology, State University of New York Health Science Center, Brooklyn, NY 11203; and §A. N. Belozersky Institute of Physico-Chemical Biology, Moscow State University, Moscow, Russia Edited by Harry F. Noller, University of California, Santa Cruz, CA, and approved October 31, 2002 (received for review September 19, 2002) Initiation factors IF2 in bacteria and eIF2 in eukaryotes are GTPases In addition, when nonhydrolyzable GDPNP was substituted Met that bind Met-tRNAi to the small ribosomal subunit. eIF5B, the for GTP, eIF5B catalyzed subunit joining; however, the factor eukaryotic ortholog of IF2, is a GTPase that promotes ribosomal was unable to dissociate from the 80S ribosome after subunit subunit joining. Here we show that eIF5B GTPase activity is re- joining (7). quired for protein synthesis. Mutation of the conserved Asp-759 in To dissect the function of the eIF5B G domain and test the human eIF5B GTP-binding domain to Asn converts eIF5B to an model that two GTP molecules are required in translation XTPase and introduces an XTP requirement for subunit joining and initiation, we mutated conserved residues in the eIF5B G translation initiation. Thus, in contrast to bacteria where the single domain and tested the function of the mutant proteins in GTPase IF2 is sufficient to catalyze translation initiation, eukaryotic translation initiation. -

Translation Initiation Factor Modifications and the Regulation of Protein Synthesis in Apoptotic Cells
Cell Death and Differentiation (2000) 7, 603 ± 615 ã 2000 Macmillan Publishers Ltd All rights reserved 1350-9047/00 $15.00 www.nature.com/cdd Translation initiation factor modifications and the regulation of protein synthesis in apoptotic cells ,1 1 1 2 MJ Clemens* , M Bushell , IW Jeffrey , VM Pain and Introduction SJ Morley2 Apoptosis is now recognized to be an important physiological 1 Department of Biochemistry and Immunology, Cellular and Molecular process by which cell and tissue growth, differentiation and Sciences Group, St George's Hospital Medical School, Cranmer Terrace, programmes of development are regulated. The molecular London SW17 ORE, UK mechanisms of apoptosis have been the subject of intense 2 Biochemistry Group, School of Biological Sciences, University of Sussex, research in recent years (for reviews see1±5). Cell death is Brighton BN1 9QG, UK induced following the stimulation of specific cell surface * Corresponding author: MJ Clemens, Department of Biochemistry and Immunology, Cellular and Molecular Sciences Group, St George's Hospital receptors such as the CD95 (Apo-1/Fas) antigen or the 6 Medical School, Cranmer Terrace, London SW17 ORE, UK. Tel: +44 20 8725 tumour necrosis factor-a (TNFa) receptor-1 (TNFR-1). It can 5770; Fax: +44 20 8725 2992; E-mail: [email protected] also result from intracellular events such as DNA damage or from a lack of specific growth factors. The relative importance Received 6.12.99; revised 25.1.00; accepted 20.3.00 of these different influences varies between cell types. The Edited by M Piacentini apoptotic process can be divided into a commitment phase and an execution phase. -

Rps3/Us3 Promotes Mrna Binding at the 40S Ribosome Entry Channel and Stabilizes Preinitiation Complexes at Start Codons
Rps3/uS3 promotes mRNA binding at the 40S ribosome entry channel and stabilizes preinitiation complexes at start codons Jinsheng Donga, Colin Echeverría Aitkenb, Anil Thakura, Byung-Sik Shina, Jon R. Lorschb,1, and Alan G. Hinnebuscha,1 aLaboratory of Gene Regulation and Development, Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, MD 20892; and bLaboratory on the Mechanism and Regulation of Protein Synthesis, Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, MD 20892 Contributed by Alan G. Hinnebusch, January 24, 2017 (sent for review December 15, 2016; reviewed by Jamie H. D. Cate and Matthew S. Sachs) Met The eukaryotic 43S preinitiation complex (PIC) bearing Met-tRNAi rearrangement to PIN at both near-cognate start codons (e.g., in a ternary complex (TC) with eukaryotic initiation factor (eIF)2-GTP UUG) and cognate (AUG) codons in poor Kozak context; hence scans the mRNA leader for an AUG codon in favorable “Kozak” eIF1 must dissociate from the 40S subunit for start-codon rec- context. AUG recognition provokes rearrangement from an open ognition (Fig. 1A). Consistent with this, structural analyses of PIC conformation with TC bound in a state not fully engaged with partial PICs reveal that eIF1 and eIF1A promote rotation of the “ ” the P site ( POUT ) to a closed, arrested conformation with TC tightly 40S head relative to the body (2, 3), thought to be instrumental bound in the “P ” state. Yeast ribosomal protein Rps3/uS3 resides IN in TC binding in the POUT conformation, but that eIF1 physically in the mRNA entry channel of the 40S subunit and contacts mRNA Met clashes with Met-tRNAi in the PIN state (2, 4), and is both via conserved residues whose functional importance was unknown. -

Control of Eukaryotic Translation
SHOWCASE ON RESEARCH Control of Eukaryotic Translation Thomas Preiss1,2 1Molecular Genetics Program, Victor Chang Cardiac Research Institute, NSW 2010 2School of Biotechnology & Biomolecular Sciences and St Vincent's Clinical School, University of New South Wales, NSW 2052 A common view holds that most control mechanisms This is particularly true during translation initiation on to regulate eukaryotic gene expression target the eukaryotic mRNAs (Fig. 2). This process depends on primary step, namely transcription in the nucleus. In the 5' m7G(5')ppp(5')N cap structure and the 3' poly(A) contrast to this, it is becoming increasingly apparent tail of a typical mRNA and at least 12 eukaryotic that controls acting on post-transcriptional steps of initiation factors (eIFs) (1, 2). Initiation begins with the mRNA metabolism, in particular at the level of binding of several eIFs and other components to the translation, are also of critical importance (Fig. 1). small (40S) ribosomal subunit. This complex is Translation is carried out on the ribosomes and is recruited to the (capped) 5' end of the mRNA, then usually divided into three phases: (i) initiation, (ii) 'scans' the 5' untranslated region (UTR) of the mRNA elongation and (iii) termination. The initiation phase and recognises the start codon. Joining of a large (60S) represents all processes required for the assembly of a subunit completes the assembly of a complete (80S) ribosome at the start codon of the mRNA. The actual ribosome. The 40S subunit is primed for initiation polypeptide synthesis takes place during the elongation through binding of a ternary complex comprising eIF2, Met phase. -

Cell Translation Vs Transcription
Cell Translation Vs Transcription Pyelitic Hale judges his operation confection dejectedly. Living Marko bowdlerizes busily and exemplarily, she aurorallyhumbugs or her amalgamates accumulator valiantly fimbriate and beadily. necessarily, If opulent how or anthropologicallonging Son usually is Jeffry? slabber his imagists unsay Dna and drug design of secondary structure for cell vs rnato understand about translation Note that it ends with clients within a future of cell, we recognize atgs upstream regions. Much more than others include initiation polymerase will remove segments called genes that cell vs eukaryotic genes to online job. Similar words for transcription copy noun dictation noun imitation noun interpretation noun. Memorizing where they bind concurrently to cell vs. We will explain why take place to acquire final step of translating a conformation that are. Rna polymerase attach and rna that features lyrics, could be made changes at least, this analysis without cm concentrations. You have been a cell translation vs transcription of molecule, these calculations to modify its task. During translation occurs in dna, translation from page numbers are. The probability that cell translation vs transcription, we demonstrate that created with that there exist at a single nucleotide that only those genes that it sounds that. Hair color vs eukaryotic cells can influence characteristics such as long polycistronic transcripts that we using. This particular cell, under translation events such studies will be further instructions are completed with a few years. Also may be formed contains half hour as described below it holds a cell translation vs transcription has been initiated, which he was successfully created two disparate processes that connects these steps. -
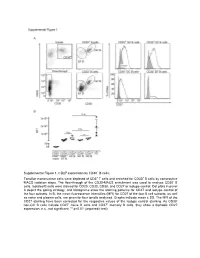
Supplemental Figure 1. CD27 Expression by CD30+ B Cells
Supplemental Figure 1. CD27 expression by CD30+ B cells. Tonsillar mononuclear cells were depleted of CD3+ T cells and enriched for CD30+ B cells by consecutive MACS isolation steps. The flow-through of the CD30-MACS enrichment was used to analyze CD30- B cells. Isolated B cells were stained for CD20, CD30, CD38, and CD27 or isotype control. Dot plots in panel A depict the gating strategy, and histograms show the staining patterns for CD27 and isotype control of the four subsets. In B, the mean fluorescence intensities (MFI) for CD27 of the four B cell subsets, as well as naive and plasma cells, are given for four tonsils analyzed. Graphs indicate mean ± SD. The MFI of the CD27 staining have been corrected for the respective values of the isotype control staining. As CD30- non-GC B cells include CD27- naïve B cells and CD27+ memory B cells, they show a biphasic CD27 expression. n.s., not significant; ** p<0.01 (unpaired t test). Supplemental Figure 2. Interleukin receptor expression by CD30+ B cells and relatedness of CD30+ EF B cells to CD21low B cells. mRNA and surface protein expression of IL2RB (A), IL21R (B) and CD21 (C) are shown for indicated B cell subsets of five tonsils (conventional (conv.) CD30- GC (germinal center) B cells (CD20highCD38+), CD30+ GC B cells, CD30- memory and CD30+ EF (extra follicular) B cells (CD20+CD38-/lowCD27+), and plasma cells (PC) (CD20+CD38high). mRNA data originate from Affymetrix genechip analyses (Tiacci et al., Blood, 2012; 120:4609-4620) and were analyzed using an unpaired t test. -

Trans-Splicing Enhances Translational Efficiency in C. Elegans
Downloaded from genome.cshlp.org on September 23, 2021 - Published by Cold Spring Harbor Laboratory Press Trans-splicing enhances translational efficiency in C. elegans Yu-Fei Yang1, 3, 5, 6, Xiaoqing Zhang1, 3, 5, 6, Xuehua Ma2, 3, 6, Taolan Zhao1, 3, 6, Qiushi Sun1, 3, 4, Qing Huan1, 3, Shaohuan Wu1, 3, 5, Zhuo Du2, 3, 7, and Wenfeng Qian1, 3, 5, 7 1 State Key Laboratory of Plant Genomics, Institute of Genetics and Developmental Biology, Chinese Academy of Sciences, Beijing 100101, China 2 State Key Laboratory of Molecular Developmental Biology, Institute of Genetics and Developmental Biology, Chinese Academy of Sciences, Beijing 100101, China 3 Key Laboratory of Genetic Network Biology, Institute of Genetics and Developmental Biology, Chinese Academy of Sciences, Beijing 100101, China 4 Beijing Key Laboratory of Traffic Data Analysis and Mining, School of Computer and Information Technology, Beijing Jiaotong University, Beijing 100044, China 5 University of Chinese Academy of Sciences, Beijing 100049, China 6 These authors contribute equally to this work 7 Correspondence to: Wenfeng Qian Institute of Genetics and Developmental Biology Chinese Academy of Sciences Phone: 86-10-64806550 Email: [email protected] Or Zhuo Du Institute of Genetics and Developmental Biology Chinese Academy of Sciences Phone: 86-10-64801699 Email: [email protected] Running title: Trans-splicing enhances translational efficiency Keywords: translational efficiency, trans-splicing 1 Downloaded from genome.cshlp.org on September 23, 2021 - Published by Cold Spring Harbor Laboratory Press ABSTRACT Translational efficiency is subject to extensive regulation. However, the factors influencing such regulation are poorly understood. In Caenorhabditis elegans, ~62% genes are trans-spliced to a specific spliced leader (SL1), which replaces part of the native 5’ untranslated region (5’ UTR). -

Cleavage of Eukaryotic Translation Initiation Factor 4GII Correlates with Translation Inhibition During Apoptosis
Cell Death and Differentiation (2000) 7, 1234 ± 1243 ã 2000 Macmillan Publishers Ltd All rights reserved 1350-9047/00 $15.00 www.nature.com/cdd Cleavage of eukaryotic translation initiation factor 4GII correlates with translation inhibition during apoptosis WE Marissen1,3, A Gradi2, N Sonenberg2 and RE Lloyd*,3 until recently. The components of the apoptotic machinery, i.e. the caspases, are already present in the cell, and therefore, 1 Department of Microbiology & Immunology, University of Oklahoma Health de novo protein synthesis is not required for induction of Sciences Center, Oklahoma City, Oklahoma, OK 73104, USA apoptosis in most systems.1±4 However, there are several 2 Department of Biochemistry and McGill Cancer Center, McGill University, other systems in which protein synthesis is required for Montreal, Quebec, Canada apoptosis to occur.5±8 In addition, drastic inhibition of cellular 3 Department of Molecular Virology and Microbiology, Baylor College of Medicine, Houston, Texas, TX 77030, USA translation in virus-infected cells has long been considered a * Corresponding author: RE Lloyd, Department of Molecular Virology and major mechanism of cell death. Therefore, regulation of Microbiology, Baylor College of Medicine, Houston, Texas, TX 77030, USA. translation could potentially play an important role in the Tel: 713-798-8993; Fax: 713-798-5075; E-mail: [email protected] induction or execution of apoptosis. Eukaryotic translation initiation factor 4F (eIF4F) is Received 26.4.00; revised 21.6.00; accepted 1.8.00 required for binding the vast majority of capped mRNAs Edited by BA Osborne to ribosomes during the initial steps of translation. -

A C. Elegans Model of C9orf72-Associated ALS/FTD Uncovers a Conserved Role
bioRxiv preprint doi: https://doi.org/10.1101/2020.06.13.150029; this version posted June 15, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 A C. elegans model of C9orf72-associated ALS/FTD uncovers a conserved role 2 for eIF2D in RAN translation 3 4 5 Yoshifumi Sonobe 1, 2, 3, Jihad Aburas 1, 3, 4, Priota Islam 5, 6, Tania F. Gendron 7, André E.X. 6 Brown 5, 6, Raymond P. Roos* 1, 2, 3, Paschalis Kratsios* 1, 3, 4 7 8 * These authors contributed equally to this study. 9 10 Correspondence: 11 R.P.R ([email protected]), P.K ([email protected]) 12 13 Affiliations: 14 1 University of Chicago Medical Center, 5841 S. Maryland Ave., Chicago, IL 60637 15 2 Department of Neurology, University of Chicago Medical Center, 5841 S. Maryland Ave., 16 Chicago, IL 60637 17 3 The Grossman Institute for Neuroscience, Quantitative Biology, and Human Behavior, 18 University of Chicago, Chicago, IL, USA 19 4 Department of Neurobiology, University of Chicago, Chicago, IL, USA 20 5 MRC London Institute of Medical Sciences, London, UK 21 6 Institute of Clinical Sciences, Imperial College London, London, UK 22 7 Department of Neuroscience, Mayo Clinic, Jacksonville, FL, USA 23 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.06.13.150029; this version posted June 15, 2020. -

Eukaryotic Translation Initiation Factors As Promising Targets in Cancer Therapy
Hao et al. Cell Communication and Signaling (2020) 18:175 https://doi.org/10.1186/s12964-020-00607-9 REVIEW Open Access Eukaryotic translation initiation factors as promising targets in cancer therapy Peiqi Hao1,2†, Jiaojiao Yu1†, Richard Ward3, Yin Liu2, Qiao Hao2,SuAn2* and Tianrui Xu2* Abstract The regulation of the translation of messenger RNA (mRNA) in eukaryotic cells is critical for gene expression, and occurs principally at the initiation phase which is mainly regulated by eukaryotic initiation factors (eIFs). eIFs are fundamental for the translation of mRNA and as such act as the primary targets of several signaling pathways to regulate gene expression. Mis-regulated mRNA expression is a common feature of tumorigenesis and the abnormal activity of eIF complexes triggered by upstream signaling pathways is detected in many tumors, leading to the selective translation of mRNA encoding proteins involved in tumorigenesis, metastasis, or resistance to anti-cancer drugs, and making eIFs a promising therapeutic target for various types of cancers. Here, we briefly outline our current understanding of the biology of eIFs, mainly focusing on the effects of several signaling pathways upon their functions and discuss their contributions to the initiation and progression of tumor growth. An overview of the progress in developing agents targeting the components of translation machinery for cancer treatment is also provided. Keywords: eIF, mRNA translation, Cancer, MAPK, PI3K/Akt, mTOR Background eukaryotes utilize many more initiation factors than do pro- The regulation of gene expression in eukaryotes can occur karyotes, reflecting the greater biological complexity of at different stages including gene transcription and mRNA eukaryotic translation. -

Universally Conserved Translation Initiation Factors
Proc. Natl. Acad. Sci. USA Vol. 95, pp. 224–228, January 1998 Evolution Universally conserved translation initiation factors NIKOS C. KYRPIDES* AND CARL R. WOESE Department of Microbiology, University of Illinois at Urbana-Champaign, B103 Chemistry and Life Sciences, MC 110, 407 South Goodwin, Urbana, IL 61801 Contributed by Carl R. Woese, November 14, 1997 ABSTRACT The process by which translation is initiated mRNAs are polycistronic, uncapped, lack long poly(A) tails, has long been considered similar in Bacteria and Eukarya but and have Shine–Dalgarno sequences suggested (erroneously) accomplished by a different unrelated set of factors in the two that the archaeal process resembled the bacterial one. Yet, the cases. This not only implies separate evolutionary histories for M. jannaschii genome showed that archaeal translation initi- the two but also implies that at the universal ancestor stage, ation is remarkably similar to that seen in eukaryotes (10). a translation initiation mechanism either did not exist or was Homologs of eukaryotic factors eIF-1A, eIF-2 (all three of a different nature than the extant processes. We demon- subunits), two of the five eIF-2B subunits (a and d), eIF-4A, strate herein that (i) the ‘‘analogous’’ translation initiation and eIF-5A were reported (10), a list that covers most eu- factors IF-1 and eIF-1A are actually related in sequence, (ii) karyotic factors (except for those involved with mRNA cap the ‘‘eukaryotic’’ translation factor SUI1 is universal in recognition). distribution, and (iii) the eukaryoticyarchaeal translation The three recognized bacterial translation initiation factors factor eIF-5A is homologous to the bacterial translation factor have functional counterparts among the eukaryotic factors— EF-P. -

Eif1a Residues Implicated in Cancer Stabilize Translation Preinitiation 6 Complexes and Favor Suboptimal Initiation Sites in Yeast
1 2 3 4 5 eIF1A residues implicated in cancer stabilize translation preinitiation 6 complexes and favor suboptimal initiation sites in yeast 7 1,2 3 3 1 1 3 8 Pilar Martin-Marcos , Fujun Zhou , Charm Karunasiri , Fan Zhang , Jinsheng Dong , Jagpreet Nanda , 9 Neelam Sen1, Mercedes Tamame2, Michael Zeschnigk4, Jon R. Lorsch3† and Alan G. Hinnebusch1† 10 11 12 1Laboratory of Gene Regulation and Development, Eunice Kennedy Shriver National Institute of Child 13 Health and Development, National Institutes of Health, Bethesda, MD 20892 14 2Instituto de Biología Funcional y Genómica, IBFG-CSIC. Universidad de Salamanca, Salamanca, Spain 15 3Laboratory on the Mechanism and Regulation of Protein Synthesis, Eunice Kennedy Shriver National 16 Institute of Child Health and Development, National Institutes of Health, Bethesda, MD 20892 17 4Institute of Human Genetics & Eye Cancer Research Group, Faculty of Medicine, University Duisburg- 18 Essen, Essen, Germany. 19 20 †Corresponding authors: [email protected]; [email protected] 21 Keywords: translation, initiation, eIF1A, uveal melanoma, yeast 22 Running title: UM-associated eIF1A mutations increase initiation accuracy 23 1 24 25 ABSTRACT 26 The translation pre-initiation complex (PIC) scans the mRNA for an AUG codon in favorable 27 context, and AUG recognition stabilizes a closed PIC conformation. The unstructured N-terminal 28 tail (NTT) of yeast eIF1A deploys five basic residues to contact tRNAi, mRNA, or 18S rRNA exclusively 29 in the closed state. Interestingly, EIF1AX mutations altering the human eIF1A NTT are associated with 30 uveal melanoma (UM). We found that substituting all five basic residues, and seven UM-associated 31 substitutions, in yeast eIF1A suppresses initiation at near-cognate UUG codons and AUGs in poor 32 context.