Evolution of Bird Genomes—
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

P0455-P0459.Pdf
The Condor 101:455-459 0 The Cooper Ornithological Society 1999 BOOK REVIEWS Social Influences on Vocal Development.- Why read the rest of the book if the main themes Charles T Snowdon and Mat-tine Hausberger, eds. are already discussed in the introduction? I believe it 1997. Cambridge University Press, Cambridge, U.K. is worth it, not only to check the statements of the ix + 351 pp. ISBN 0521 49526 1. $95.00 (cloth). editors. All the articles are fine reviews of a given At least in certain aspects, this book is a referee’s field, written by experts. Doug Nelson provides a very dream: not only do the editors provide short summaries good account of the state of the art in song learning in their introduction of the 16 articles making up this theory, and as I expressed above, I fully agree with his volume, they also inform the reader about the main claim that the two stage nature of song learning is still general conclusions that can be drawn from those ar- not fully understood or accepted by other researchers ticles. According to Snowdon and Hausberger, there of the field. are five central themes that arc treated in almost all Luis Baptista and Sandra Gaunt provide a very com- contributions. The first and main theme is the claim petent review on social interaction and vocal devel- that vocal learning has more aspects than learning to opment in birds. As ever, there is a wealth of findings produce sounds. It is also necessary to learn how to from the lab and from the field included in their re- use and comprehend vocalizations. -
1 ID Euring Latin Binomial English Name Phenology Galliformes
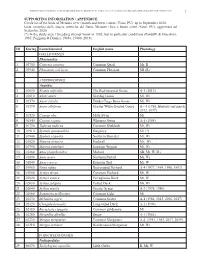
BIRDS OF METAURO RIVER: A GREAT ORNITHOLOGICAL DIVERSITY IN A SMALL ITALIAN URBANIZING BIOTOPE, REQUIRING GREATER PROTECTION 1 SUPPORTING INFORMATION / APPENDICE Check list of the birds of Metauro river (mouth and lower course / Fano, PU), up to September 2020. Lista completa delle specie ornitiche del fiume Metauro (foce e basso corso /Fano, PU), aggiornata ad Settembre 2020. (*) In the study area 1 breeding attempt know in 1985, but in particolar conditions (Pandolfi & Giacchini, 1985; Poggiani & Dionisi, 1988a, 1988b, 2019). ID Euring Latin binomial English name Phenology GALLIFORMES Phasianidae 1 03700 Coturnix coturnix Common Quail Mr, B 2 03940 Phasianus colchicus Common Pheasant SB (R) ANSERIFORMES Anatidae 3 01690 Branta ruficollis The Red-breasted Goose A-1 (2012) 4 01610 Anser anser Greylag Goose Mi, Wi 5 01570 Anser fabalis Tundra/Taiga Bean Goose Mi, Wi 6 01590 Anser albifrons Greater White-fronted Goose A – 4 (1986, february and march 2012, 2017) 7 01520 Cygnus olor Mute Swan Mi 8 01540 Cygnus cygnus Whooper Swan A-1 (1984) 9 01730 Tadorna tadorna Common Shelduck Mr, Wi 10 01910 Spatula querquedula Garganey Mr (*) 11 01940 Spatula clypeata Northern Shoveler Mr, Wi 12 01820 Mareca strepera Gadwall Mr, Wi 13 01790 Mareca penelope Eurasian Wigeon Mr, Wi 14 01860 Anas platyrhynchos Mallard SB, Mr, W (R) 15 01890 Anas acuta Northern Pintail Mi, Wi 16 01840 Anas crecca Eurasian Teal Mr, W 17 01960 Netta rufina Red-crested Pochard A-4 (1977, 1994, 1996, 1997) 18 01980 Aythya ferina Common Pochard Mr, W 19 02020 Aythya nyroca Ferruginous -

Leptosomiformes ~ Trogoniformes ~ Bucerotiformes ~ Piciformes
Birds of the World part 6 Afroaves The core landbirds originating in Africa TELLURAVES: AFROAVES – core landbirds originating in Africa (8 orders) • ORDER ACCIPITRIFORMES – hawks and allies (4 families, 265 species) – Family Cathartidae – New World vultures (7 species) – Family Sagittariidae – secretarybird (1 species) – Family Pandionidae – ospreys (2 species) – Family Accipitridae – kites, hawks, and eagles (255 species) • ORDER STRIGIFORMES – owls (2 families, 241 species) – Family Tytonidae – barn owls (19 species) – Family Strigidae – owls (222 species) • ORDER COLIIFORMES (1 family, 6 species) – Family Coliidae – mousebirds (6 species) • ORDER LEPTOSOMIFORMES (1 family, 1 species) – Family Leptosomidae – cuckoo-roller (1 species) • ORDER TROGONIFORMES (1 family, 43 species) – Family Trogonidae – trogons (43 species) • ORDER BUCEROTIFORMES – hornbills and hoopoes (4 families, 74 species) – Family Upupidae – hoopoes (4 species) – Family Phoeniculidae – wood hoopoes (9 species) – Family Bucorvidae – ground hornbills (2 species) – Family Bucerotidae – hornbills (59 species) • ORDER PICIFORMES – woodpeckers and allies (9 families, 443 species) – Family Galbulidae – jacamars (18 species) – Family Bucconidae – puffbirds (37 species) – Family Capitonidae – New World barbets (15 species) – Family Semnornithidae – toucan barbets (2 species) – Family Ramphastidae – toucans (46 species) – Family Megalaimidae – Asian barbets (32 species) – Family Lybiidae – African barbets (42 species) – Family Indicatoridae – honeyguides (17 species) – Family -

Wqfm: Statistically Consistent Genome-Scale Species Tree Estimation from Weighted Quartets
bioRxiv preprint doi: https://doi.org/10.1101/2020.11.30.403352; this version posted December 1, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. wQFM: Statistically Consistent Genome-scale Species Tree Estimation from Weighted Quartets Mahim Mahbub1;y, Zahin Wahab1;y, Rezwana Reaz1;y, M. Saifur Rahman1, and Md. Shamsuzzoha Bayzid1,* 1Department of Computer Science and Engineering Bangladesh University of Engineering and Technology Dhaka-1205, Bangladesh yThese authors contributed equally to this work *Corresponding author: shams [email protected] Abstract Motivation: Species tree estimation from genes sampled from throughout the whole genome is complicated due to the gene tree-species tree discordance. Incom- plete lineage sorting (ILS) is one of the most frequent causes for this discordance, where alleles can coexist in populations for periods that may span several speciation events. Quartet-based summary methods for estimating species trees from a collec- tion of gene trees are becoming popular due to their high accuracy and statistical guarantee under ILS. Generating quartets with appropriate weights, where weights correspond to the relative importance of quartets, and subsequently amalgamating the weighted quartets to infer a single coherent species tree allows for a statistically consistent way of estimating species trees. However, handling weighted quartets is challenging. Results: We propose wQFM, a highly accurate method for species tree es- timation from multi-locus data, by extending the quartet FM (QFM) algorithm to a weighted setting. -

Birds of Madagascar and Their Conservation
Birds of Madagascar and Their Conservation byMichael S. Putnam Department ofZoology University of Wisconsin tl Madison, Wisconsin I 100 plus Ecstatic Testimonials Striding up a steep hillside with the Council for Bird Preservation (Collar loud whisper of a rushing stream in and Stuart, 1985), 28 species of Warm, nurturing foods. the background, I stepped into a Malagasy birds are threatened and 14 mist-net lane I had cut a week before. species are considered as near Cook monthly, As I entered the clearing, a medium threatened. These species represent freeze in packets, sized brown bird squawked and flew between one-fifth and one-third of off from eye-level. Carefully search the island's endemic bird species. serve in seconds. ing the nearby vegetation, I became one of a lucky handful of foreigners The primary threats to Madagas At fine stores, orcall... to ever find a nest of the Brown car's birds today, habitat loss and Mesite (Mesitornis unicolor), a rare overhunting, have already eliminated 1(800) BIRD YUM forest-dwelling relative of rails. The many unique Malagasy creatures. 1 (800) 247-3986 large egg, delicately colored in sal Since people first arrived on Mada mon with liver-colored spots, rested gascar 1500 to 2000 years ago, much 13330 Bessemer Street precariously in a frail, dove-like nest of the island has been deforested, Van Nuys, CA91401-3000 positioned at the end of a sloping leaving the red lateritic soil exposed (818) 997-0598 sapling. This encounter with the and eroding, with little chance for Brown Mesite is just one of many forest regeneration. -

Journal of Avian Biology JAV-00869 Wang, N
Journal of Avian Biology JAV-00869 Wang, N. and Kimball, R. T. 2016. Re-evaluating the distribution of cooperative breeding in birds: is it tightly linked with altriciality? – J. Avian Biol. doi: 10.1111/jav.00869 Supplementary material Appendix 1. Table A1. The characteristics of the 9993 species based on Jetz et al. (2012) Order Species Criteria1 Developmental K K+S K+S+I LB Mode ACCIPITRIFORMES Accipiter albogularis 0 0 0 0 1 ACCIPITRIFORMES Accipiter badius 0 0 0 0 1 ACCIPITRIFORMES Accipiter bicolor 0 0 0 0 1 ACCIPITRIFORMES Accipiter brachyurus 0 0 0 0 1 ACCIPITRIFORMES Accipiter brevipes 0 0 0 0 1 ACCIPITRIFORMES Accipiter butleri 0 0 0 0 1 ACCIPITRIFORMES Accipiter castanilius 0 0 0 0 1 ACCIPITRIFORMES Accipiter chilensis 0 0 0 0 1 ACCIPITRIFORMES Accipiter chionogaster 0 0 0 0 1 ACCIPITRIFORMES Accipiter cirrocephalus 0 0 0 0 1 ACCIPITRIFORMES Accipiter collaris 0 0 0 0 1 ACCIPITRIFORMES Accipiter cooperii 0 0 0 0 1 ACCIPITRIFORMES Accipiter erythrauchen 0 0 0 0 1 ACCIPITRIFORMES Accipiter erythronemius 0 0 0 0 1 ACCIPITRIFORMES Accipiter erythropus 0 0 0 0 1 ACCIPITRIFORMES Accipiter fasciatus 0 0 0 0 1 ACCIPITRIFORMES Accipiter francesiae 0 0 0 0 1 ACCIPITRIFORMES Accipiter gentilis 0 0 0 0 1 ACCIPITRIFORMES Accipiter griseiceps 0 0 0 0 1 ACCIPITRIFORMES Accipiter gularis 0 0 0 0 1 ACCIPITRIFORMES Accipiter gundlachi 0 0 0 0 1 ACCIPITRIFORMES Accipiter haplochrous 0 0 0 0 1 ACCIPITRIFORMES Accipiter henicogrammus 0 0 0 0 1 ACCIPITRIFORMES Accipiter henstii 0 0 0 0 1 ACCIPITRIFORMES Accipiter imitator 0 0 0 0 1 ACCIPITRIFORMES -

Attempting to See One Member of Each of the World's Bird Families Has
Attempting to see one member of each of the world’s bird families has become an increasingly popular pursuit among birders. Given that we share that aim, the two of us got together and designed what we believe is the most efficient strategy to pursue this goal. Editor’s note: Generally, the scientific names for families (e.g., Vireonidae) are capital- ized, while the English names for families (e.g., vireos) are not. In this article, however, the English names of families are capitalized for ease of recognition. The ampersand (&) is used only within the name of a family (e.g., Guans, Chachalacas, & Curassows). 8 Birder’s Guide to Listing & Taxonomy | October 2016 Sam Keith Woods Ecuador Quito, [email protected] Barnes Hualien, Taiwan [email protected] here are 234 extant bird families recognized by the eBird/ Clements checklist (2015, version 2015), which is the offi- T cial taxonomy for world lists submitted to ABA’s Listing Cen- tral. The other major taxonomic authority, the IOC World Bird List (version 5.1, 2015), lists 238 families (for differences, see Appendix 1 in the expanded online edition). While these totals may appear daunting, increasing numbers of birders are managing to see them all. In reality, save for the considerable time and money required, finding a single member of each family is mostly straightforward. In general, where family totals or family names are mentioned below, we use the eBird/Clements taxonomy unless otherwise stated. Family Feuds: How do world regions compare? In descending order, the number of bird families supported by con- tinental region are: Asia (125 Clements/124 IOC), Africa (122 Clem- ents/126 IOC), Australasia (110 Clements/112 IOC), North America (103 Clements/IOC), South America (93 Clements/94 IOC), Europe (73 Clements/74 IOC ), and Antarctica (7 Clements/IOC). -

A Community Effort to Annotate the Chicken Genome
bioRxiv preprint doi: https://doi.org/10.1101/012559; this version posted December 12, 2014. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Submitted to Cytogenetic and Genome Research as part of the Third Report on Chicken Genes and Chromosomes The Avian RNAseq Consortium: a community effort to annotate the chicken genome (Prepared by Jacqueline Smith, David W. Burt and the Avian RNAseq Consortium) Publication of the chicken genome sequence in 2004 (International Chicken Genome Sequencing Consortium 2004) highlighted the beginning of a revolution in avian genomics. Progression of DNA sequencing technologies and data handling capabilities has also meant that genome sequencing and assembly is now a relatively simple, fast and inexpensive procedure. The success seen with the chicken genome was soon followed by the completion of the zebra finch genome (Warren et al., 2010), an important model for neurobiology (Clayton et al., 2009), again based on Sanger sequencing. In recent years the rapid advances in Next Generation Sequencing (NGS) technologies, hardware and software have meant that many more genomes can now be sequenced faster and cheaper than ever before (Metzker, 2010). The first avian genome to be sequenced by NGS methods was the turkey (Dalloul et al., 2010), which was also integrated with genetic and physical maps thus providing an assembly of high quality, even at the chromosome level. Recently, NGS has been used to sequence the genomes of a further 42 avian species, as part of the G10K initiative (Genome 10K Community of Scientists, 2009). -

Columbiformes ~ Pterocliformes ~ Mesitornithiformes
Birds of the World part 2 Galloanseres, Neoaves: Columbea NEOGNATHAE (the rest of the birds!): Galloanseres • ORDER ANSERIFORMES – waterfowl • Family Anhimidae – screamers (3 species) • Family Anseranatidae – magpie goose (1 species) • Family Anatidae – ducks, geese, and swans (173 species) • ORDER GALLIFORMES – landfowl • Family Megapodiidae – megapodes (21 species) • Family Cracidae – chachalacas, curassows, and guans (55 species) • Family Numididae – guineafowl (6 species) • Family Odontophoridae – New World quail (34 species) • Family Phasianidae – pheasants and allies (183 species) NEOGNATHAE : Neoaves (the rest of the birds!): COLUMBEA • ORDER PODICIPEDIFORMES – Family Podicipedidae – grebes (23 species) • ORDER PHOENICOPTERIFORMES – Family Phoenicopteridae – flamingos (6 species) • ORDER COLUMBIFORMES – Family Columbidae – pigeons and doves (334 species) • ORDER PTEROCLIDIFORMES – Family Pteroclididae – sandgrouse (16 species) • ORDER MESITORNITHIFORMES – Family Mesitornithidae – mesites (3 species) NEOGNATHAE : Galloanseres • ORDER ANSERIFORMES – waterfowl • Family Anhimidae – screamers (3 species) • Family Anseranatidae – magpie goose (1 species) • Family Anatidae – ducks, geese, and swans (173 species) • ORDER GALLIFORMES – landfowl • Family Megapodiidae – megapodes (21 species) • Family Cracidae – chachalacas, curassows, and guans (55 species) • Family Numididae – guineafowl (6 species) • Family Odontophoridae – New World quail (34 species) • Family Phasianidae – pheasants and allies (183 species) southern or crested screamer -

Data Types and the Phylogeny of Neoaves
Article Data Types and the Phylogeny of Neoaves Edward L. Braun * and Rebecca T. Kimball * Department of Biology, University of Florida, Gainesville, FL 32611, USA * Correspondence: ebraun68@ufl.edu (E.L.B.); rkimball@ufl.edu (R.T.K.) Simple Summary: Some of the earliest studies using molecular data to resolve evolutionary history separated birds into three main groups: Paleognathae (ostriches and allies), Galloanseres (ducks and chickens), and Neoaves (the remaining ~95% of avian species). The early evolution of Neoaves, however, has remained challenging to understand, even as data from whole genomes have become available. We have recently proposed that some of the conflicts among recent studies may be due to the type of genomic data that is analyzed (regions that code for proteins versus regions that do not). However, a rigorous examination of this hypothesis using coding and non-coding data from the same genomic regions sequenced from a relatively large number of species has not yet been conducted. Here we perform such an analysis and show that data type does influence the methods used to infer evolutionary relationships from molecular sequences. We also show that conducting analyses using models of sequence evolution that were chosen to minimize reconstruction errors result in coding and non-coding trees that are much more similar, and we add to the evidence that non-coding data provide better information regarding neoavian relationships. While a few relationships remain problematic, we are approaching a good understanding of the evolutionary history for major avian groups. Abstract: The phylogeny of Neoaves, the largest clade of extant birds, has remained unclear despite intense study. -

Silvery-Cheeked Hornbill Bycanistes Brevis
Silvery-cheeked Hornbill Bycanistes brevis Class: Aves Order: Bucerotiformes Family: Bucerotidae Characteristics: A fairly large bird with a cream-colored, lightweight casque (“horn”) on top of its beak. The head has silvery-grey feathers (hence the name) and the rest of the body is covered with black or white feathers. In males, the casque is larger than the females. Otherwise they are quite similar. Behavior: They usually live in pairs and, as they are fairly gregarious, will sometimes roost in flocks of several hundred individuals (Beauty of Birds). They have a fairly wide range through which they fly long distances feeding on fruits and small birds in flight. They return to their tree roost at night. Reproduction: They breed in the African spring (September and October). The female finds a cavity and, with the male’s help, seals herself in leaving a small hole through which he feeds her regurgitated fruit. She lays a clutch of 1-3 eggs which hatch following a 40-day incubation period (Oiseaux Birds). Diet: Wild: mainly fruits but will also eat insects, small birds, rodents, small reptiles, centipedes Zoo: softbill, fruit/veg mix, hardboiled egg, dog food and parrot pellets Conservation: The population appears to be quite stable and has a large range, however their numbers seem to be declining in Zimbabwe (Biodiversity Explorer). FYI: The hornbill is one of the few birds that have eyelashes to shield them from sun and dust (Pittsburg Zoo). . -

Preliminary Survey on Relative Diversity and Residential Status of Avifauna in District Karnal, Haryana ( India)
International Journal of Advanced Scientific Research and Management, Volume 4 Issue 6, June 2019 www.ijasrm.com ISSN 2455-6378 Preliminary Survey on Relative Diversity and Residential Status of Avifauna in district Karnal, Haryana ( India). Parveen Kumar Vats Associate Professor, Department of Zoology,Pt. C.L.S. Govt. College Karnal Karnal, Haryana, India Abstract transferring nutrients and spores from one place to The present study was conducted in another during their migration and local movements different sites of district Karnal, Haryana (India) (Niemi, 1985). Birds show variations greatly in their from May 2017 to April 2019. A total of 122 bird diversity, habitats, abundance and distribution species were observed by using line and point count throughout the globe. They have usually more method. These 122 bird species belongs to 17 diversity in tropics than others regions like different orders and maximum bird species were temperate, alpine or polar. Their habitat preferences observed in order Passeriformes (48) followed by are more or less specialized. They occupy higher order Pelecaniformes (14). Accipitridae and trophic levels in food webs. They also vary in their Ardeidae were two families in which equal number abundance because some species occur in large of bird species (8) were observed. A total of 88 bird numbers while others are represented by few species out of 122 observed avian species were individuals only. Some avian species have small resident, 29 bird species were winter migrants and 5 breeding ranges in some particular region whereas bird species were observed only in summer during others travel long distance and annual migrations the present study.