Direct Amide Formation from Unactivated Carboxylic Acids and Amines
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

02/06/2019 12:05 PM Appendix 3745-21-09 Appendix A
ACTION: Final EXISTING DATE: 02/06/2019 12:05 PM Appendix 3745-21-09 Appendix A List of Organic Chemicals for which Paragraphs (DD) and (EE) of Rule 3745-21-09 of the Administrative Code are Applicable Organic Chemical Organic Chemical Acetal Benzaldehyde Acetaldehyde Benzamide Acetaldol Benzene Acetamide Benzenedisulfonic acid Acetanilide Benzenesulfonic acid Acetic acid Benzil Acetic Anhydride Benzilic acid Acetone Benzoic acid Acetone cyanohydrin Benzoin Acetonitrile Benzonitrile Acetophenone Benzophenone Acetyl chloride Benzotrichloride Acetylene Benzoyl chloride Acrolein Benzyl alcohol Acrylamide Benzylamine Acrylic acid Benzyl benzoate Acrylonitrile Benzyl chloride Adipic acid Benzyl dichloride Adiponitrile Biphenyl Alkyl naphthalenes Bisphenol A Allyl alcohol Bromobenzene Allyl chloride Bromonaphthalene Aminobenzoic acid Butadiene Aminoethylethanolamine 1-butene p-aminophenol n-butyl acetate Amyl acetates n-butyl acrylate Amyl alcohols n-butyl alcohol Amyl amine s-butyl alcohol Amyl chloride t-butyl alcohol Amyl mercaptans n-butylamine Amyl phenol s-butylamine Aniline t-butylamine Aniline hydrochloride p-tertbutyl benzoic acid Anisidine 1,3-butylene glycol Anisole n-butyraldehyde Anthranilic acid Butyric acid Anthraquinone Butyric anhydride Butyronitrile Caprolactam APPENDIX p(183930) pa(324943) d: (715700) ra(553210) print date: 02/06/2019 12:05 PM 3745-21-09, Appendix A 2 Carbon disulfide Cyclohexene Carbon tetrabromide Cyclohexylamine Carbon tetrachloride Cyclooctadiene Cellulose acetate Decanol Chloroacetic acid Diacetone alcohol -

Amide, and Paratoluenesulfonamide on the Amide of Silver,On the Imides
68 CHEMISTRY: E. C. FRANKLIN METALLIC SALTS OF AMMONO ACIDS By Edward C. Franklin DEPARTMENT OF CHEMISTRY, STANFORD UNIVERSITY Presented to the Academy, January 9. 1915 The Action of Liquid-Ammonia Solutions of Ammono Acids on Metallic Amides, Imides, and Nitrides. The acid amides and imides, and the metallic derivatives of the acid amides and imides are the acds, bases, and salts respectively of an ammonia system of acids, bases, and salts.1 Guided by the relationships implied in the above statement Franklin and Stafford were able to prepare potassium derivatives of a considerable number of acid amides by the action of potassium amide on certain acid amides in solution in liquid ammonia. That is to say, an ammono base, potassium amide, was found to react with ammono acids in liquid ammonia to form ammono salts just as the aquo base, potassium hydrox- ide, acts upon aquo acids in water solution to form aquo salts. Choos- ing, for example, benzamide and benzoic acid as representative acids of the two systems, the analogous reactions taking place respectively in liquid ammonia and water are represented by the equations: CH6CONH2+KNH2 = C6H5CONHK + NHs. CseHCONH2 + 2KNH2 = CIHsCONK2 + 2NH3. CH6tCOOH + KOH = CIH6COOK + H2O. The ammono acid, since it is dibasic, reacts with either one or two molecules of potassium amide to form an acid and a neutral salt. Having thus demonstrated the possibility of preparing ammono salts of potassium by the interaction of potassium amide and acid amides in liquid ammonia solution, it was further found that ammono salts of the heavy metals may be prepared by the action of liquid ammonia solutions of ammono acids on insoluble metallic amides, imides, and nitrides-that is, by reactions which are analogous to the formation of aquo salts in water by the action of potassium hydroxide on insoluble metallic hydroxides and oxides. -

Amide Activation: an Emerging Tool for Chemoselective Synthesis
Featuring work from the research group of Professor As featured in: Nuno Maulide, University of Vienna, Vienna, Austria Amide activation: an emerging tool for chemoselective synthesis Let them stand out of the crowd – Amide activation enables the chemoselective modification of a large variety of molecules while leaving many other functional groups untouched, making it attractive for the synthesis of sophisticated targets. This issue features a review on this emerging field and its application in total synthesis. See Nuno Maulide et al., Chem. Soc. Rev., 2018, 47, 7899. rsc.li/chem-soc-rev Registered charity number: 207890 Chem Soc Rev View Article Online REVIEW ARTICLE View Journal | View Issue Amide activation: an emerging tool for chemoselective synthesis Cite this: Chem. Soc. Rev., 2018, 47,7899 Daniel Kaiser, Adriano Bauer, Miran Lemmerer and Nuno Maulide * It is textbook knowledge that carboxamides benefit from increased stabilisation of the electrophilic carbonyl carbon when compared to other carbonyl and carboxyl derivatives. This results in a considerably reduced reactivity towards nucleophiles. Accordingly, a perception has been developed of amides as significantly less useful functional handles than their ester and acid chloride counterparts. Received 27th April 2018 However, a significant body of research on the selective activation of amides to achieve powerful DOI: 10.1039/c8cs00335a transformations under mild conditions has emerged over the past decades. This review article aims at placing electrophilic amide activation in both a historical context and in that of natural product rsc.li/chem-soc-rev synthesis, highlighting the synthetic applications and the potential of this approach. Creative Commons Attribution 3.0 Unported Licence. -

Reactions of Benzene & Its Derivatives
Organic Lecture Series ReactionsReactions ofof BenzeneBenzene && ItsIts DerivativesDerivatives Chapter 22 1 Organic Lecture Series Reactions of Benzene The most characteristic reaction of aromatic compounds is substitution at a ring carbon: Halogenation: FeCl3 H + Cl2 Cl + HCl Chlorobenzene Nitration: H2 SO4 HNO+ HNO3 2 + H2 O Nitrobenzene 2 Organic Lecture Series Reactions of Benzene Sulfonation: H 2 SO4 HSO+ SO3 3 H Benzenesulfonic acid Alkylation: AlX3 H + RX R + HX An alkylbenzene Acylation: O O AlX H + RCX 3 CR + HX An acylbenzene 3 Organic Lecture Series Carbon-Carbon Bond Formations: R RCl AlCl3 Arenes Alkylbenzenes 4 Organic Lecture Series Electrophilic Aromatic Substitution • Electrophilic aromatic substitution: a reaction in which a hydrogen atom of an aromatic ring is replaced by an electrophile H E + + + E + H • In this section: – several common types of electrophiles – how each is generated – the mechanism by which each replaces hydrogen 5 Organic Lecture Series EAS: General Mechanism • A general mechanism slow, rate + determining H Step 1: H + E+ E El e ctro - Resonance-stabilized phile cation intermediate + H fast Step 2: E + H+ E • Key question: What is the electrophile and how is it generated? 6 Organic Lecture Series + + 7 Organic Lecture Series Chlorination Step 1: formation of a chloronium ion Cl Cl + + - - Cl Cl+ Fe Cl Cl Cl Fe Cl Cl Fe Cl4 Cl Cl Chlorine Ferric chloride A molecular complex An ion pair (a Lewis (a Lewis with a positive charge containing a base) acid) on ch lorine ch loronium ion Step 2: attack of -

United States Patent Office Patented May 16, 1967 2 3,320,314 125 C
3,320,314 United States Patent Office Patented May 16, 1967 2 3,320,314 125 C. Agitation may be employed during the reaction, CHLOROBENZYL SULFAMDES but none is required. William J. Houlihan, Mountain Lakes, N.J., assignor to The tertiary amine medium provides a solvent system Sandoz Inc., Hanover, N.J. in which the reaction takes place. Contemplated tertiary No Drawing. Filled June 15, 1964, Ser. No. 375,288 5 amines include, for example, tri(lower) alkylamines, e.g. 6 Claims. (C. 260-556) triethylamine; (lower) alkyl pyrroles, e.g. N-propyl-pyr This application is a continuation-in-part of application role; pyridine; (lower) alkyl pyridines, e.g. 3-ethyl pyri Ser. No. 339,354, filed on Jan. 22, 1964, and now aban dine; (lower) alkoxy pyridines, e.g. 2,5-dimethoxypyri doned. dine; quinoline; (lower) alkyl quinolines, e.g. 8-ethyl O quinoline; N-(lower) alkyl morpholine, e.g. N-methyl This invention is directed to two groups of benzyl sulf morpholine; and N,N'-di(lower) alkyl piperazine, e.g. amides having one or more chlorine substituents on the N-methyl,N'-ethyl-piperazine. sole aromatic ring. These groups are, respectively, of For the preparation of Compounds II wherein R6 is a the formulae hydrogen atom similar reaction conditions are employed; R1 a primary benzyl amine (V) is substituted for the sec ondary benzyl amine (III), and the reaction medium is an R2- ceN seNH aqueous ethanolic medium: R R3- -H 20 R1 R4 (I) and R2- ot NH R1 -- (IV) - (II) -- NH3 R3 -Rs R' ce ise 25 R2- N NEI k (B) R6 R3 -R5 (V) RA (II) 30 wherein In both reaction (A) and reaction (B) each of R, R, R is either lower alkyl having two or more carbon atoms R, R3, R, R5 and R6 has its above-ascribed meaning. -

Hydrogenation of Benzonitrile Over Supported Pd Catalysts: Kinetic and Mechanistic Insight Mairi I
This is an open access article published under a Creative Commons Attribution (CC-BY) License, which permits unrestricted use, distribution and reproduction in any medium, provided the author and source are cited. Article Cite This: Org. Process Res. Dev. 2019, 23, 977−989 pubs.acs.org/OPRD Hydrogenation of Benzonitrile over Supported Pd Catalysts: Kinetic and Mechanistic Insight Mairi I. McAllister,† Cedrić Boulho,† Lauren F. Gilpin,† Liam McMillan,† Colin Brennan,‡ and David Lennon*,† † School of Chemistry, Joseph Black Building, University of Glasgow, Glasgow G12 8QQ, U.K. ‡ Syngenta, Jeallot’s Hill International Research Centre, Berkshire RG42 6EY, U.K. ABSTRACT: The liquid phase hydrogenation of benzonitrile over a 5 wt % Pd/C catalyst using a stirred autoclave is investigated. The reaction conforms to a consecutive reaction sequence: first benzonitrile is hydrogenated to produce benzylamine, which subsequently undergoes a hydrogenolysis step to form toluene. Benzonitrile hydrogenation obeys first-order kinetics with an activation energy of 27.6 kJ mol−1. In contrast, the benzylamine hydrogenolysis stage obeys zero-order kinetics −1 and exhibits an activation energy of 80.1 kJ mol . A 1 wt % Pd/Al2O3 catalyst is additionally examined, which is also seen to support hydrogenolysis activity alongside the hydrogenation pathway. Gas phase transmission infrared spectroscopic measurements of the hydrogenation of benzonitrile and benzylamine over the 1 wt % Pd/Al2O3 catalyst utilizing hydrogen and deuterium are undertaken, which enable reaction schemes incorporating adsorption geometries of intermediate adsorption complexes to be proposed. KEYWORDS: nitrile hydrogenation, carbon-supported palladium, hydrogenolysis, benzonitrile, benzylamine 1. INTRODUCTION selectivity (95%).12 The high selectivity toward the primary Primary aromatic amines represent an important class of amine was obtained under mild conditions (303 K, 6 bar H2) chemicals with widespread application in many areas of the in the presence of an acid additive. -

Further Studies on the Synthesis Of
FURTHER STUDIES ON THE SYNTHESIS OF ARYLETHMOLMINES By Robert Simonoff in Thesis submitted to the Faculty of the Graduate School of the University of Maryland in partial fulfillment of the requirements for the degree of Doctor of Philosophy 1945 UMI Number: DP70015 All rights reserved INFORMATION TO ALL USERS The quality of this reproduction is dependent upon the quality of the copy submitted. In the unlikely event that the author did not send a complete manuscript and there are missing pages, these will be noted. Also, if material had to be removed, a note will indicate the deletion. UMI Dissertation Publishing UMI DP70015 Published by ProQuest LLC (2015). Copyright in the Dissertation held by the Author. Microform Edition © ProQuest LLC. All rights reserved. This work is protected against unauthorized copying under Title 17, United States Code ProQuest ProQuest LLC. 789 East Eisenhower Parkway P.O. Box 1346 Ann Arbor, Ml 48106- 1346 ACKNOWLEDGEMENT The author wishes to express his appreciation for the encouragement and assistance given by Dr, Walter H. Hartung under whose direction this work has been carried out* TABLE OF CONTENTS Page INTRODUCTION.................................................... 1 REVIEW OF THE LITERATURE Previous Methods of Synthesis of Arylethanolamines Hydrogenolytic Debenzylation. ................. ......17 EXPERIMENTAL Synthesis of Ketones .................... .33 Synthesis of Amines.......... ........ ......... ....... ....... 38 Nitrosation of Ketones.• •«••••••.......... 40 Decomposition of Arylglyoxylohydroxamyl -
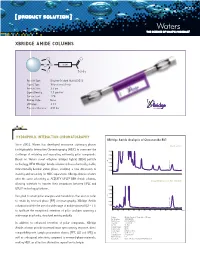
Xbridge Amide Columns
[ PRODUCT SOLUTION ] XBRIDGE AMIDE COLUMNS O O O Si Linker O NH 2 Particle Type: Ethylene Bridged Hybrid [BEH] Ligand Type: Trifunctional Amide Particle Size: 3.5 µm Ligand Density: 7.5 µmol/m2 Carbon Load: 12% Endcap Style: None pH Range: 2-11 Pressure Tolerance: 400 bar HYDROPHILIC INTERacTION CHROMATOGRAPHY XBridge Amide Analysis of Ginsenoside Rb1 Since 2003, Waters has developed innovative stationary phases Orento extract for Hydrophilic Interaction Chromatography [HILIC] to overcome the 0.010 challenge of retaining and separating extremely polar compounds. 0.008 Based on Waters novel ethylene bridged hybrid [BEH] particle 0.006 AU technology, NEW XBridge™ Amide columns utilize a chemically stable, 0.004 0.002 Ginsenoside Rb1 trifunctionally-bonded amide phase, enabling a new dimension in 0.000 stability and versatility for HILIC separations. XBridge Amide columns offer the same selectivity as ACQUITY UPLC® BEH Amide columns, 20 µg/mL Ginsenoside Rb1 standard allowing scientists to transfer their separations between HPLC and 0.010 ® UPLC technology platforms. 0.008 0.006 Designed to retain polar analytes and metabolites that are too polar AU 0.004 to retain by reversed-phase [RP] chromatography, XBridge Amide 0.002 columns facilitate the use of a wide range of mobile phase pH [2 – 11] 0.000 Ginsenoside Rb1 to facilitate the exceptional retention of polar analytes spanning a 0.00. 5 1.01. 5 2.02. 5 3.03. 5 4.04. 5 5.05. 5 6.06. 5 min wide range in polarity, structural moiety and pKa. Column: XBridge Amide, 3.5 µm, 4.6 x 150 mm Part Number: 186004869 Mobile Phase : 80/20 ACN/H2O In addition to enhanced retention of polar compounds, XBridge Flow Rate: 1.4 mL/min Inj. -

Bis(Benzylamine) Monomers: One-Pot Preparation and Application in Dendrimer Scaffolds for Removing Pyrene from Aqueous Environments
Supporting Information for Bis(benzylamine) monomers: One-pot preparation and application in dendrimer scaffolds for removing pyrene from aqueous environments Olivia N. Monaco, Sarah C. Tomas, Meghan K. Kirrane and Amy M. Balija* Address: Department of Chemistry, Fordham University, 441 E. Fordham Road, Bronx, NY 10458, United States Email: Amy M. Balija* - [email protected] *Corresponding author Full experimental synthetic procedures for compounds 3a–g, 4a–h,6–8, and 11–13, and pyrene fluorescence spectral data Table of contents: I. General experimental II. Synthesis of bisimine compounds 3 III. Synthesis of bisamine compounds 4 IV. Synthesis of dendrimers 6, 11–13 V. Thin film fluorescence experiments VI. Calculations of inclusion formation constants, Gibbs free energies, and dendrimer capacities VII. Representative 1H and 13C NMR spectral data VIII. References I. General experimental All reactions were performed under an argon gas atmosphere with either flame-dried or oven-dried glassware unless otherwise noted. Reagents were obtained from Aldrich or TCI America. 2-(Dimethylamino)pyridinium p-toluenesulfonate (DPTS) was synthesized as reported previously [1]. Solvents and reagents were used without further purification except for the following: MeOH was distilled from CaSO4, CH2Cl2 was distilled from CaH2, benzaldehyde was distilled neat, and phloroglucinol dihydrate was azeotroped 5 times with toluene prior to use. Reactions were monitored by thin layer chromatography (TLC) using silica gel 60 F254 glass plates. TLC bands were visualized by UV and phosphomolybdic acid (PMA) stain. Eluent solvent ratios are reported in v/v. Size exclusion chromatography was performed using a 2 cm x 50 cm column of Bio-Rad Bio- Beads S-X1 beads (200–400 mesh) in toluene. -

COMMUNICATION Catalytic Ester and Amide To
Catalytic Ester and Amide to Amine Interconversion: Nickel-Catalyzed Decarbonylative Amination of Esters and Amides by C−O and C−C Bond Activation Item Type Article Authors Yue, Huifeng; Guo, Lin; Liao, Hsuan-Hung; Cai, Yunfei; Zhu, Chen; Rueping, Magnus Citation Yue H, Guo L, Liao H-H, Cai Y, Zhu C, et al. (2017) Catalytic Ester and Amide to Amine Interconversion: Nickel-Catalyzed Decarbonylative Amination of Esters and Amides by C−O and C −C Bond Activation. Angewandte Chemie International Edition 56: 4282–4285. Available: http://dx.doi.org/10.1002/anie.201611819. Eprint version Post-print DOI 10.1002/anie.201611819 Publisher Wiley Journal Angewandte Chemie International Edition Rights This is the peer reviewed version of the following article: Catalytic Ester and Amide to Amine Interconversion: Nickel-Catalyzed Decarbonylative Amination of Esters and Amides by C-O and C-C Bond Activation, which has been published in final form at http:// doi.org/10.1002/anie.201611819. This article may be used for non-commercial purposes in accordance With Wiley Terms and Conditions for self-archiving. Download date 23/09/2021 09:53:43 Link to Item http://hdl.handle.net/10754/623218 COMMUNICATION Catalytic Ester and Amide to Amine Interconversion: Nickel- Catalyzed Decarbonylative Amination of Esters and Amides via C- O and C-C Bond Activation Huifeng Yue[a], Lin Guo[a], Hsuan-Hung Liao[a], Yunfei Cai[a], Chen Zhu[a], and Magnus Rueping[a,b]* Abstract: An efficient nickel catalyzed decarbonylative amination reaction of aryl and heteroaryl esters has been achieved for the first a) Schmidt reaction; Curtius, Hoffmann, Lossen rearrangement time. -

Dissociation Constants of Organic Acids and Bases
DISSOCIATION CONSTANTS OF ORGANIC ACIDS AND BASES This table lists the dissociation (ionization) constants of over pKa + pKb = pKwater = 14.00 (at 25°C) 1070 organic acids, bases, and amphoteric compounds. All data apply to dilute aqueous solutions and are presented as values of Compounds are listed by molecular formula in Hill order. pKa, which is defined as the negative of the logarithm of the equi- librium constant K for the reaction a References HA H+ + A- 1. Perrin, D. D., Dissociation Constants of Organic Bases in Aqueous i.e., Solution, Butterworths, London, 1965; Supplement, 1972. 2. Serjeant, E. P., and Dempsey, B., Ionization Constants of Organic Acids + - Ka = [H ][A ]/[HA] in Aqueous Solution, Pergamon, Oxford, 1979. 3. Albert, A., “Ionization Constants of Heterocyclic Substances”, in where [H+], etc. represent the concentrations of the respective Katritzky, A. R., Ed., Physical Methods in Heterocyclic Chemistry, - species in mol/L. It follows that pKa = pH + log[HA] – log[A ], so Academic Press, New York, 1963. 4. Sober, H.A., Ed., CRC Handbook of Biochemistry, CRC Press, Boca that a solution with 50% dissociation has pH equal to the pKa of the acid. Raton, FL, 1968. 5. Perrin, D. D., Dempsey, B., and Serjeant, E. P., pK Prediction for Data for bases are presented as pK values for the conjugate acid, a a Organic Acids and Bases, Chapman and Hall, London, 1981. i.e., for the reaction 6. Albert, A., and Serjeant, E. P., The Determination of Ionization + + Constants, Third Edition, Chapman and Hall, London, 1984. BH H + B 7. Budavari, S., Ed., The Merck Index, Twelth Edition, Merck & Co., Whitehouse Station, NJ, 1996. -

Use of Chemically Modified Poly(Ethylene Terephthalate)-G- (Acryl Amide) Fibers for Α-Amylase Immobilization
http://www.e-polymers.org e-Polymers 2007, no. 070 ISSN 1618-7229 Use of chemically modified poly(ethylene terephthalate)-g- (acryl amide) fibers for α-amylase immobilization Zülfikar Temoçin, Mustafa Yiğitoğlu* Kırıkkale University, Science and Arts Faculty, Chemistry Department, Yahşihan, 71450 Kırıkkale, Turkey; Fax: +90-318-3572461; [email protected]. (Received: 12 April, 2007; published: 30 June, 2007) Abstract: Acryl amide grafted Poly(ethylene terephthalate) (AAm-g-PET) fiber was used for covalent coupling of α-amylase. The amide groups of Poly(acryl amide) were converted to the amine groups by Hofmann degradation reaction. The amine groups were activated by glutaraldehyde, before coupling of the enzyme. The free α-amylase and immobilized α-amylase were characterized by determining the activity profile as function of pH, temperature, thermal stability and storage stability. For the immobilized α-amylase, operational stability was also determined. The immobilization of α-amylase on support caused the optimal reaction pH to shift from 5 to 6. The maximum activity of the free and immobilized enzymes occurred at 0 50 C. Km for the immobilized system was higher than that for the free enzyme. The activity of the free enzyme ended in 30 days, whereas the activity of the immobilized enzyme lasted for 60 days at storage conditions. α-Amylase immobilized on matrix maintained 40% of its original activity after 30 times of repeated use. Introduction α-Amylase is a kind of starch-hydrolyzed enzyme. It is an endo-enzyme that can cut α-1,4 glucosidic linkage of starch molecules randomly to form oligo with different molecular weight such as glucose, cane sugar and straight chain oligo, rapidly reducing the viscosity of liquid and has been used in big volume starch hydrolysis industry [1].