Dr. Meena Patel Dr. Bharat Pateliya* Dr. Khushbu K. Tilva
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
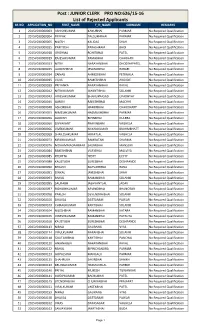
JUNIOR CLERK PRO NO:626/15-16 List of Rejected Applicants
Post : JUNIOR CLERK PRO NO:626/15-16 List of Rejected Applicants SR.NO APPLICATION_NO FIRST_NAME F_H_NAME SURNAME REMARKS 1 2015V030000003 ASHISHKUMAR KANUBHAI PARMAR No Required Qualification 2 2015V030000004 DEEPAK DALSUKHBHAI PARMAR No Required Qualification 3 2015V030000005 NILESH KALIDAS SHAH No Required Qualification 4 2015V030000015 PARITOSH PARASHRAM BHOI No Required Qualification 5 2015V030000018 GRISHMA ROHITBHAI PATEL No Required Qualification 6 2015V030000019 RAJESHKUMAR PAMABHAI CHAUHAN No Required Qualification 7 2015V030000023 NITIN NARAYANBHAI DHODIYAPATEL No Required Qualification 8 2015V030000025 JAGDISHBHAI JESANGBHAI RABARI No Required Qualification 9 2015V030000034 ZAINAB AHMEDIBHAI PETIWALA No Required Qualification 10 2015V030000035 VILAS RAMESHBHAI ARGADE No Required Qualification 11 2015V030000039 PRIYANKA NARAYANBHAI RAVAL No Required Qualification 12 2015V030000042 NITIN KUMAR JAYANTIBHAI SOLANKI No Required Qualification 13 2015V030000043 VINESHKUMAR BHANUPRASAD UPADHYAY No Required Qualification 14 2015V030000045 JAIMIN RAKESHBHAI MACHHI No Required Qualification 15 2015V030000048 ASHOKBHAI HAMIRBHAI CHAUDHARY No Required Qualification 16 2015V030000055 NIMESHKUMAR HASMUKHBHAI PARMAR No Required Qualification 17 2015V030000056 JAGDISH BIPINBHAI DULERA No Required Qualification 18 2015V030000060 DIVYAKANT PRAVINBHAI VAGHELA No Required Qualification 19 2015V030000066 VIVEKKUMAR NAYANKUMAR BRAHMBHATT No Required Qualification 20 2015V030000069 SHAILESHKUMAR HIMATLAL VAGHELA No Required Qualification 21 2015V030000073 -

Dr. Pravin R. Prajapati
Dr. Pravin R. Prajapati ..... CAREER OBJECTIVE Contact 3, Umiya Nagar To be in the field of academics as a teacher, where I can use my passion to B/h 22 Gam School develop young minds by empowering them with necessary skills, qualities, Opp. Krishna Housing knowledge, attitude and value of time. To be part of a progressive organi- Sco., zation that gives scope to enhance my knowledge, skills and to reach the Vallabh Vidyanagar pinnacle in the research field with sheer determination, dedication and hard Gujarat -388120 work. India +91 9429367045 +91 8791633827 EDUCATION 2012 - 2015 PhD RF & Microwave Engg. [8.00 CPI out of 10] Indian Institute of Technology, ⃝ pravinprajapati05 a Roorkee gmail.com IIT-Roorkee 2004 - 2006 M Tech Communication Systems [8.52 CPI out of 10] Indian Institute of www.prprajapati.co.in Technology, Varanasi Modeling and Banaras Hindu University, Varanasi (IT-BHU) 1991 - 1993 12th. [71%] St. Xavier’s High School, Anand, Gujarat Analysis Gujarat Higher Education Board, Gujarat CST Microwave Studio Optsim 1990 - 1991 10th. [81%] St. Xavier’s High School, Anand, Gujarat Matlab Gujarat Higher Education Board, Gujarat Software & Tools EXPERIENCE MS Office 2006 to till today Associate Professor A. D. Patel Institute of Technology, Gujarat MS Visio Latex 2002 to 2006 Lecturer A. D. Patel Institute of Technology, Gujarat Academic Identity 2001 to 2002 Lecturer U. V. Patel College of Engineering, Mehsana, Gujarat Google Scholar ID: p2u6u5sAAAAJ 2001-2001 Visiting Lecturer Birla Vishwakarma Mahavidyalaya (Engg. College, Gujarat) Researcher ID: C-2315-2015 1999-2000 Adhoc Lecturer Birla Vishwakarma Mahavidyalaya (Engg. College, Gujarat) Scopus ID: 54403556900 Orcid ID: 0000-0001-7455-4194 Vidwan ID:123471 LABORATORIES CONDUCTED Indexes Fiber Optic Communication Technology Communication Techniques Wireless Commu- Google Scholar nication Biomedical Instrumentation Electronics Projects I &II Bio-Medical Engg. -

Deepayan Nayak
This list includes all the JNV students (from all States and Union Territories) invited to take the Navodaya Dakshana Selection Test 2020 (NDST) on Sunday, December 08, 2019. (Last update: November 19, 2019) Following 19 JNVs are not included in the list: Class 10 Result not received: Indore, Anantnag, Budgam, Baramulla, Ganderbal, Kulgam, Kupwara, Rajouri, Shopian, Car Nicobar, Farrukhabad, Ghaziabad School Don’t have Class 10: Sirmour, Rohtas, Howrah, Jalpaiguri, Nandurbar – II, Saiha, Kiphire Page 1 of 192 Attn. Principals and Students: Only students listed in this document can appear for the Navodaya Dakshana Selection Test – NDST 2020. Principals are requested not to send anyone else as the test centers have capacity constraints and they will not be permitted to take the test. IMPORTANT: All students whose name appears in the NDST Selection List MUST fill the Dakshana Scholar Application Form online latest by November 25, 2019. Invited students who do not register will not be eligible to sit for the JDST. 1. The online application form is be available at http://dakshana.org/become-a-scholar/ 2. Please fill the form online and submit. 3. Admit Card can be downloaded from December 01, 2019. 4. Affix photograph and have the Admit Card signed by your school Principal. How to fill Online Scholar Application Form: To apply visit: http://scholarship.dakshana.org/ For Detailed Instruction on how to fill Online Dakshana Scholar Application form: https://tinyurl.com/dak2020 Step 1: Log In: Enter your Dakshana Roll No. Now enter your Date of Birth which will be your password. (The format of the date of birth to be entered is “YYYY-MM-DD”. -

This List Includes All the JNV Students (From All States and Union Territories) Invited to Take the Joint Dakshana Selection
This list includes all the JNV students (from all States and Union Territories) invited to take the Joint Dakshana Selection Test 2020 (JDST) on Sunday, December 08, 2019. (Last update: December 04, 2019) Following JNVs are not included in the list: Result not received: 03 JNV Rajouri, Farrukhabad, Arwal Page 1 of 156 Schools don’t have Class 12 Science: 54 JNV Sukma, Jhabua - (St), Malkangiri – II, Lahaul & Spiti, Kulgam, Car Nicobar, Banswara - II (St), Sri Ganganagar – II, Deoria, Sitapur II, Jehanabad, Khagaria, Sheohar, Gaya - II (Sc), Palamu – I, Pakur - II (St), Palamu - II (Sc), Kooch Bihar, Darjeeling, Howrah, Jalpaiguri, South 24 Paraganas – I, Uttar Dinajpur, North Goa, Dang, Narmada, Dhule, Nandurbar – II, Anjaw, East Siang, Khurung Khumey, Papumpare, Upper Dibang Valley, Upper Siang, West Kameng, Dima Hasao (N. C. Hills), Tamenglong, Senapati II, Ukhrul II, East Khasi Hills - II (St), Champhai, Kolasib, Lunglei, Mamit, Saiha, Dimapur, Kiphire, Longleng, Mokokchung, Phek, Tuensang, Wokha, Zunheboto, East Sikkim Attn. Principals and Students: Only students listed in this document can appear for the Joint Dakshana Selection Test – JDST 2020. Principals are requested not to send anyone else as the test centers have capacity constraints and they will not be permitted to take the test. IMPORTANT: All students whose name appears in the JDST Selection List MUST fill the Dakshana Scholar Application Form online latest by November 25, 2019. Invited students who do not register will not be eligible to sit for the JDST. 1. The online application form is be available at http://dakshana.org/become-a-scholar/ 2. Please fill the form online and submit. -

NORTHWESTERN UNIVERSITY Unearthing Subaltern Agency: The
NORTHWESTERN UNIVERSITY Unearthing Subaltern Agency: The Representation of Marginalized Populations in Contemporary Indian Literature A DISSERTATION SUBMITTED TO THE GRADUATE SCHOOL IN PARTIAL FULFILMENT OF THE REQUIREMENTS for the degree DOCTOR OF PHILOSOPHY Field of English By Tanushree Vachharajani EVANSTON, ILLINOIS September 2017 2 © Copyright by Tanushree Vachharajani 2017 All Rights Reserved 3 Acknowledgements I am deeply grateful for the members of my dissertation committee who have been extraordinarily supportive during my writing process: to Dr. Evan Mwangi, my advisor, whose insight into how gaps in my argument could be filled has made my dissertation richer, and who comforted me during moments of panic during my dissertation writing; to Dr. Dilip Gaonkar, whose classes and work introduced me to the field of diaspora studies and with whom I got the chance to meet many new thinkers; to Dr. Andrew Leong who taught me to deconstruct, restructure and sharpen my argument and to respond to academic audiences; and to Dr. Laura Brueck, who taught me the importance of the fields of translation and Dalit studies, and whose work closely informs my own. I would also like to thank Mr. Dalpat Chauhan, a prolific Gujarati Dalit writer and the editor of Vanboti Vartao. He has been particularly helpful with the niceties of the translation process and with putting me in touch with the authors of the stories. Finally, I would like to thank my family – my husband, Tanay, my sister Vidisha and my parents Narendra and Yamini. They have been a source of great strength and support through many trying moments during my PhD. -

Development of Low Cost Protection Technology for Shoot Fly and Stem Borer in Pearl Millet Crop
Journal of Entomology and Zoology Studies 2021; 9(1): 1562-1565 E-ISSN: 2320-7078 P-ISSN: 2349-6800 Development of low cost protection technology for www.entomoljournal.com JEZS 2021; 9(1): 1562-1565 shoot fly and stem borer in pearl millet crop © 2021 JEZS Received: 25-10-2020 Accepted: 22-12-2020 RP Juneja, GM Parmar, KD Mungra, Asha C Detroja, DL Kadvani and RP Juneja SK Parmar Pearl Millet Research Station, Junagadh Agriculture University, Jamnagar, Gujarat, DOI: https://doi.org/10.22271/j.ento.2021.v9.i1v.8360 India Abstract GM Parmar Field experiment was conducted at Pearl Millet Research Station, Junagadh Agricultural University, Pearl Millet Research Station, Jamnagar during Kharif 2016 to 2018 to find out the effective and economical control measures against Junagadh Agriculture shoot fly and stem borer in pearl millet crop. The results showed that the seed treatment of clothianidin University, Jamnagar, Gujarat, 50 WDG @ 7.5 g/kg seed followed by spray of fipronil 5 SC @ 0.01% at 35 days after germination was India found effective against shoot fly. Whereas, the seed treatment of clothianidin 50 WDG @ 7.5 g/kg seed KD Mungra followed by spray of chlorantraniliprole 20 SC @ 0.006% at 35 days after germination was found Pearl Millet Research Station, effective against stem borer. The highest additional income (Rs. 17940/-) and net return (Rs. 15975/-) Junagadh Agriculture was recorded with the seed treatment of clothianidin 50 WDG @ 7.5 g/kg seed followed by spray of University, Jamnagar, Gujarat, fipronil 5 SC @ 0.01% at 35 days after germination. -

Appeal Coordinating Office
150 route de Ferney, P.O. Box 2100 1211 Geneva 2, Switzerland Tel: 41 22 791 6033 Fax: 41 22 791 6506 e-mail: [email protected] Appeal Coordinating Office India Emergency Drought Relief ASIN-02 Appeal Target: US$ 1,083,684 Geneva, 12 May 2000 Dear Colleagues, A severe drought has gripped large parts of the Near East and South Asia. The affected region cuts like an east - west swathe from Orissa to Rajasthan through central India and beyond through Baluchistan and Sindh in Pakistan, south and central Afghanistan and into Iran, Iraq and Jordan. The UN’s Food and Agriculture Organization reports that it is part of the wider climatic phenomenon that has also adversely affected a number of countries in northern and eastern Africa. In India alone, more than 100 million people are affected by the drought, which has cast its shadow over 12 states. The worst affected states are Rajasthan, Gujarat, Madhya Pradesh, Andhra Pradesh and Orissa. ACT members, Church’s Auxiliary for Social Action (CASA) and Lutheran World Service (LWS) India have visited the affected region and met with local partners as well as with UN, NGO and government officials. The following proposals to provide much needed relief assistance to the most vulnerable sections of the distressed population are a result of the visits and meetings. ACT is a worldwide network of churches and related agencies meeting human need through coordinated emergency response. The ACT Coordinating Office is based with the World Council of Churches (WCC) and the Lutheran World Federation (LWF) in Switzerland. -

Government of Rajasthan
Government of Rajasthan POST ENUMERATION SURVEY OF 5 PERCENT SAMPLE CHECKING OF DISE DATA IN THREE DISTRICTS OF CHURU, DUNGARPUR & JHALAWAR IN RAJASTHAN: 2010-11 DIRECTORATE OF ECONOMICS AND STATISTICS, GOVERNMENT OF RAJASTHAN, YOJANA BHAWAN, JAIPUR 2011 2 Directorate of Economics and Statistics, Rajasthan POST ENUMERATION SURVEY OF 5 PERCENT SAMPLE CHECKING OF DISE DATA IN THREE DISTRICTS OF CHURU, DUNGARPUR & JHALAWAR IN RAJASTHAN: 2010-11 Submitted to Department of School Education, Government of Rajasthan DIRECTORATE OF ECONOMICS AND STATISTICS, GOVERNMENT OF RAJASTHAN, YOJANA BHAWAN, JAIPUR 2011 3 Directorate of Economics and Statistics, Rajasthan POST ENUMERATION SURVEY OF 5 PERCENT SAMPLE CHECKING OF DISE DATA IN THREE DISTRICTS OF CHURU, DUNGARPUR & JHALAWAR IN RAJASTHAN: 2010-11 Submitted to Department of School Education, Government of Rajasthan DIRECTORATE OF ECONOMICS AND STATISTICS, GOVERNMENT OF RAJASTHAN, YOJANA BHAWAN, JAIPUR 2011 4 Directorate of Economics and Statistics, Rajasthan T A B L E O F C O N T E N T S Sl. No. Contents Page No. 1.0 Preface …………………………………………………………………………… 6 2.0 List of Figures…………………………………………………………………… 7 3.0 List of Tables……………………………………………………………………. 8 4.0 Abbreviations …………………………………………………………………. 13 5.0 Executive Summary………………………………………………………….. 14 6.0 Chapter 1 : Introduction……………………………………………………. 23 6.1 The Status of Literacy and Education in India and Rajasthan………… 25 6.2 Socio-demographic Profile of Rajasthan & Districts……………………… 30 6.3 Status of Literacy and Education in Rajasthan and Districts……….... 32 6.4 Objectives of the Study………………………………………………………… 40 7.0 Chapter 2 : Study Methodology…………………………………………… 41 7.1 Study Design…………………………………………………………………….. 42 7.2 Sampling Procedure……………………………………………………………. 42 7.3 Data Collection………………………………………………………………….. 42 7.4 Field Operations and Supervision…………………………………………… 43 7.5 Scrutiny and Validation………………………………………………………. -

1. District Brief Profile
1. DISTRICT BRIEF PROFILE A- DISTRICT POLITICAL MAP B- KEY STATISTICS POPULATION – 2011 1241519 PROJECTED POPULATION - (Current Year) 1464992 POPULATION DENSITY 194 LITERACY RATE TOTAL 63.23% LITERACY RATE MALE 74.05% LITERACY RATE FEMALE 51.36% GROWTH RATE – TOTAL 26.97 GROWTH RATE – URBAN 30.87 GROWTH RATE – RURAL 25.71 NO. OF MUNICIPAL CORPORATION 00 NO. OF MUNICIPALITIES 05 NO. OF BLOCKS 05 NO. OF GRAM PANCHAYATS 425 NO. OF REVENUE VILLAGES 1375 TOTAL ELECTORS IN DISTRICT 815962 MALE ELECTORS 431346 FEMALE ELECTORS 384553 SEX RATIO – 2011 911 SEX RATIO - (As per final roll) EP RATIO – 2011 EP RATIO - (As per final roll) NO. OF PARLIAMENTARY CONSTITUENCIES (PC) 02 (Partially) NO. OF POLING STATION (PS) 1065 NO. OF POLLING STATION LOCATIONS 970 NO. OF ASSEMBLY CONSTITUENCIES (AC) 04 NO. OF RETURNING OFFICERS (RO) 04 NO. OF AROS 12 NO. OF SECTOR OFFICERS (SO) NO. OF BLOS 1065 C- BRIEF NOTIES ON THE DISTRICT Write very briefly about the district India is a vast country and considering the size, geography and culture, conducting election has always been an arduous task. It is termed as a biggest festival of democracy, whereby, a government is elected through the people’s participation. It is indeed a tough task for the authority to conduct the free and fair polling with proper security to electors and polling personnel. Hence, it is strongly recommended to have a robust election management plan, in line with the prescribed guidelines and instructions, with the authority which deals with the each and every aspect of election preparedness. All officers are, therefore, advised to be familiarized and acquainted with each measure to be adopted at various stages of electoral processes and election management plan can be a better tool to guide the concerned officials. -
Some Wild Vegetable Plant Used by Tribals of Dhar District, Madhya Pradesh
Indian J. Applied & Pure Bio. Vol. 31(1), 65-69 (2016). Ethnobotany: Some wild Vegetable Plant used by Tribals of Dhar District, Madhya Pradesh K.S. Alawa and Sudip Ray Department of Botany, Govt. P.G. College, Dhar (India) Department of Botany, PMB Gujarati Science College, Indore (India) Email: [email protected] and [email protected] Abstract The tribals living in remote forest areas with respected to food plants showed that tribals depend much upon forest products for their eaten a variety of wild vegetable plants. The paper deals with communicates first-hand information on 32 plant species belonging to 31 genera and 23 families were collected and identified. The information is based on an ethnobotanical field study of the district during 2012 to 2015. The tribals depend much upon forest products for their varies daily needs. The botanical names are arranged in alphabetical order, vernacular name and families, along with their plant part used and method to preparation is discussed. In all 32 plant species such utilized of vegetable, viz. Abelmoschus ficulneus (L.)Wt., Amaranthus spinosa L., Argyreia nervosa (Burm.f.)Bojer., Bauhinia racemosa Lam.,Celosia argentea L.,Corchorus olitorius L., Cordia dichotoma Forst. f., Momordica dioica Roxb.ex Willd., Senna tora (L.) Roxb., Wattakaka volubilis (L.f.) Stapf. were observred. Dhar district is situated in the south- the forest and largely dependent on the wild western part of Madhya Pradesh, India. The biological resources for their livelihood. They study area lies between 22º oo to 23º 10 utilize wide variety of plant for their basic needs Northern latitude and 74º 28 to 75º 42 Eastern such as food, fodder, fiber, wood, medicine, longitude. -
Article Download
wjpls, 2019, Vol. 5, Issue 3, 140-142 Research Article ISSN 2454-2229 Alawa . World Journal of Pharmaceutical World Journal and Life of Pharmaceutical Sciences and Life Sciences WJPLS www.wjpls.org SJIF Impact Factor: 5.008 ETHNOBOTANICAL OBSERVATION ON MUSICAL INSTRUMENT USED BY TRIBALS OF DHAR DISTRICT, MADHYA PRADESH, INDIA AND THEIR ROLE IN CONSERVATION Dr. Kamal Singh Alawa* Assistant Professor, Department of Botany, Govt. P.G. College, Dhar, (M.P.) India. *Corresponding Author: Dr. Kamal Singh Alawa Assistant Professor, Department of Botany, Govt. P.G. College, Dhar, (M.P.) India. Article Received on 28/12/2018 Article Revised on 18/01/2019 Article Accepted on 08/02/2019 ABSTRACT Ethnobotanical some plants identification on musical instruments during 2017 to 2018. The paper deals with communicates first information on 14 plant species belonging to 14 families were collected and identified in the study area. The main tribes Bheel and Sub-tribe Bhilala, Barela and Pateliya are the dominant tribes living in remote areas. Tribals are very fond of simple formulations of wisdom acquired over long periods of enjoying to song and music. Music is an art form, social or cultural activity whose medium is sound and silence. The common musical instruments like “Dhol, Mandal” Dholak, Dhak and “Flute” or “Pouli’ are used in classical music. In this paper we have studied for musical instruments that appears to human ears as similar with respect to music plants and their role in conservation. KEYWORDS: Ethnobotany, Musical instruments, Festivals, Madhya Pradesh, Tribals. INTRODUCTION Kuralkar et al. 2015; Alawa et al. 2012, 2016, 2018 & Alawa 2018). -
Green India Mission Perspective Plan (Revised)
Green India Mission Perspective Plan (Revised) 2016-17 to 2020-21 MP FOREST DEPARTMENT Index 1 Chapter 1-Introduction ................................................................................ 1.1. About Madhya Pradesh.............................................................................................................. 3 1.2. Ecological Importance of Madhya Pradesh in Central Indian Landscape ........................... 3 1.3. Forest in Madhya Pradesh ......................................................................................................... 4 1.4. Wild Life of Madhya Pradesh .................................................................................................... 4 1.5. Wild Life Corridors in Madhya Pradesh: ................................................................................ 5 1.6. Biosphere Reserves in MadhyaPradesh: .................................................................................. 6 1.7. Major River Basins of Madhya Pradesh .................................................................................. 7 1.8. Waste Lands in Madhya Pradesh .............................................................................................. 8 1.9. Demography ................................................................................................................................ 9 1.10. Dependence on Forests: ............................................................................................................ 10 1.11. Joint Forest Management .......................................................................................................