Matinas Biopharma Holdings, Inc. 10K 2020 V1
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Life Science District
Fitzsimons Life Science District a great place to raise an idea Located in Aurora, Colorado, it’s one of the largest bioscience developments in the country, a place where there’s plenty of room to grow an idea from discovery to market viability. Whether you start in the bioscience incubator or move into a state-of-the art lab facility, everything you need is close at hand. A cluster of talent and resources is available to you, including direct access to the more than 80 core laboratories at the University of Colorado Anschutz Medical Campus – all just steps away. Features throughout the district, such as parks and conference facilities, are designed to inspire collaboration. And a nearby mix of shops, restaurants and homes let your people thrive right along with your ideas. For leasing and land purchase opportunities at the Colorado Science + Technology Park, within the Fitzsimons Life Science District, call 720-941-7100. Join the conversation at FitzScience.com FIT 110352 Brand Ad_M.indd 1 3/23/11 1:32 PM Improve the health of your business. Collaborations between bioscience 227 acres of state-of-the-art patient successful Colorado companies such companies and faculty researchers at care, research and education, the as Myogen/ Gilead Sciences Colorado, the University of Colorado Anschutz Anschutz Medical Campus provides GlobeImmune, ARCA, and ApopLogic Medical Campus are flourishing. unique opportunities for business Pharmaceuticals. And with Denver Which means the opportunities for development. The University’s International Airport being only 15 improvement—in the health of Technology Transfer o ce, ranked minutes away, access to both coasts Coloradoans and in the health of among the top 20 universities for takes only a few hours. -

ЗМШИ ВНЕСЕНО Наказ Y1ihicerepctba Охорони Здоров'я Укратни
ЗАТВЕРДЖЕНО Наказ MiHic•cpcTBa охорони здоров'я УкраТни ЗМШИ ВНЕСЕНО Наказ Y1iHicerepcTBa охорони здоров'я УкраТни 1 Н СТРУКЦIЯ для медичного застосування засобу БЮВЕН (BIOVEN) СктаД: ДЈючаречовина: Нитап normal immunoglobulin for intravenous administration; мл препарату активноТ 6iJIk0B0T G —(),l г; Допомјжнј речовини: (кислота вода для форма. Розчин для ОсновнЈ властивостј.• прозора або з незначното опалесцен[јею, безбарвна або злегка жовтуватого кольору Фармакотерапевтична трупа. людини нормальний для введення. код АТХ Ј06В А02. властивостј. Препарат е активною G у IgGl: 52 %, lgG2: 32 %, IgG3: 7 %, IgG4: 4 %), граничний BMiCT А у становить 400 мкг/мл. компонентом препарату е що akTIIBHicT10проти захворювань —BipyciB i у т.ч. гепатиту А i В, Bipycy герпеса людини 1 типу, 2 типу та 6 типу, Bipycy Епштейна-Барр, грипу, кору, паротиту, краснухи, коклюшу, кишковоТ налички, пневмокока, правцевого та токсину. Мас також що проявлясгься у Препарат низькою спонтанною антикомплементарною Препарат с нативним О, BCi комплементу, ефекторну та опсоно-фагоцитарну Препаратс активною 6iJIk0B010 що з сироватки або плазми kPOBi людини, на до BlJl-l, до Bipycy гепатиту С та поверхневого антигену Bipycy гепатиту В, очищеною та концентрованою методом спиртоводними осадниками, яка пройшла BipycH0T сольвент-детергентним методом. про модельних BipyciB у табл. 1. Таблиця BipyciB Результат випробування Bipyc Фактор титру методомПЛР Bi с iM оде людини 5.01 ТСП)ю/см Bi с гепап С 5,51 Bi сп стогоге пес 11-готип 6,01 ТСП)5Ысм Bipyc BipycH0T великоТ рога- 5,5 тот х доби Bi с псевдосказ 10ТСЮ5Ысм Енте Bi с свиней 1-го тип 4,6 1 с людини У-го тип 10ТСП)$Ысм Bi с гепа каченят 1-го тип н/д Bi с вези ля ного стомати 7,01 10 н/д ж/д —немае даних Фарл:акокгнетика. -

2020 Ncbiotech Company Directory Lists 735 Life Science Companies Across the State
NCBIOTECH Company Directory 2020 NC’s 35-Year Transformation When civic and business leaders decided in 1984 that North Carolina should take a leadership role in staking an economic future in the life sciences, they started by establishing the North Carolina Biotechnology Center. At the time, there were 32 companies statewide that could be considered part of that sector, employing 3,000 people. Now, after 35 years of continued state investment in the Center, the print version of this 2020 NCBiotech Company Directory lists 735 life science companies across the state. And today they employ 66,000 people in a wide range of excellent jobs that pay, on average, twice the average private-sector wage. Then there are 2,400 more support companies, listed in the online version of this directory. In all, the sector directly or indirectly impacts some 240,000 people statewide. It all adds up to $83.3 billion in annual economic activity for North Carolina, and an influx of $2.2 billion in state and local tax income. Of the 735 companies in this directory, 160 of them are based on technologies developed at North Carolina universities. One of the most successful, started as Quintiles (now IQVIA) in a tiny mobile office on the campus of the University of North Carolina at Chapel Hill, has given birth to the multi-billion-dollar Contract Research and Testing industry. North Carolina remains the epicenter of the field, now home to more than 150 such companies employing 24,250 workers. NCBiotech has been the constant catalyst through all this growth, deploying billions of dollars in research funding to universities across the state, and, through the years, $44 million in loans to 211 entrepreneurial companies. -
Immunogenicity and Safety of Fractional Doses of Yellow Fever Vaccines: a Randomized
medRxiv preprint doi: https://doi.org/10.1101/2020.08.18.20177527; this version posted October 29, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. It is made available under a CC-BY-NC-ND 4.0 International license . 1 Immunogenicity and safety of fractional doses of yellow fever vaccines: a randomized, 2 double-blind, non-inferiority trial 3 4 Aitana Juan-Giner, MSc1,*, Derick Kimathi, MBChB2,3*, Kyra H. Grantz, BA4,5,6, Mainga M. 5 Hamaluba, MD2, Patrick Kazooba, MBChB7, Patricia Njuguna, MMed2, Gamou Fall, PhD8, 6 Moussa Dia, MSc8, Ndeye S. Bob, PhD8, Thomas P. Monath, MD9, Alan D. Barrett, PhD10, 7 Joachim Hombach, PhD11, Edgar M. Mulogo, PhD12, Immaculate Ampeire, MBChB13, Henry 2 7 7 8 K. Karanja, MSc , Dan Nyehangane, MSc , Juliet Mwanga-Amumpaire, MMed , Derek A.T. 9 Cummings, PhD4,5, Philip Bejon, Prof2,3, George M. Warimwe, PhD2,3,#, Rebecca F. Grais, 10 PhD1,#. 11 12 1 Epicentre, Paris, France 13 2 Kenya Medical Research Institute – Wellcome Trust Research Programme, Kilifi, Kenya 14 3 Centre for Tropical Medicine & Global Health, University of Oxford, UK 15 4 Department of Biology, University of Florida 16 5 Emerging Pathogens Institute, University of Florida 17 6 Department of Epidemiology, Johns Hopkins Bloomberg School of Public Health 18 7 Epicentre Mbarara Research Centre, Uganda 19 8 Institut Pasteur Dakar, Senegal 20 9 Crozet BioPharma LLC, Devens MA, USA 21 10 Sealy Institute for Vaccines Sciences and Department of Pathology, University of Texas 22 Medical Branch, USA 23 11 Immunization, Vaccines & Biologicals, World Health Organization, Geneva, Switzerland 24 12 Department of Community Health, Mbarara University of Science & Technology, Uganda 25 13 Expanded Program on Immunization, Ministry of Health, Uganda 26 27 *Co-first author; #Co-senior author 28 1 NOTE: This preprint reports new research that has not been certified by peer review and should not be used to guide clinical practice. -

Annual Report 2020
ANNUAL REPORT 2020 Caring for People’s Health as a Trusted Partner 2020 A Year like none before Peter Goldschmidt CEO Dear Investors, dear Partners, 2020 was for each of us a demanding year. Like all of you, we at STADA had to deal with new challenges and to adapt to a new situation in our jobs as well as at home with our family. I would like to express my gratitude to all employees worldwide. You have made an extraordinary contribution to our society through your tireless efforts. We can be proud of the strong crisis resilience STADA has shown despite challeng- ing market conditions during the pandemic. The company has proven itself to be a growth leader and go-to-partner in Consumer Healthcare and Generics, with a rapidly expanding Specialty Pharma presence in areas such as Parkinson’s and biosimilars. I’m particularly gratified by how our diverse global team has come through the crisis true to our Purpose of “Caring for People’s Health as a Trusted Partner”. Across the organization, people have pulled together, even if they have been physically distant; this close collaboration across continents truly embodies our value One STADA. Keep Supplying Medicines We care for people’s health as a trusted partner by being able to keep supplying medicines, even during lockdowns. Our 6,000 production staff did an outstanding job in these difficult times. STADA provides a broad portfolio from supplements that enable people to stay healthy and support their immune systems through to essential treatments for diseases in nearly every therapeutic area, such as cancer, heart, central nervous system, gastrointestinal and pain relief. -

Company Directory
NCBIOTECH Company Directory ncbiotech.org/directory NC’s Life Science Transformation The 34 years of work by the North Carolina Biotechnology Center has proven to be transformative for the state – especially for its manufacturing workforce. As employment in the state’s furniture, textile and tobacco factories tumbled over the past three decades, the number of people working in pharmaceutical and medicine manufacturing has grown from a few dozen to about 21,000 today. And pharmaceutical manufacturing is just one important segment of North Carolina’s transformational life science community. It doesn’t include the thousands of people in high-tech agriculture, or medical device commercialization, or contract research and development – a life science ecosystem totaling 63,000-plus workers at more than 700 companies statewide. The annual NCBiotech Company Directory print edition aggregates information on these 700+ companies, while the online version includes nearly 2,300 supporting companies such as patent law firms specializing in life science work. They bring the direct and indirect life science workforce total to 260,000. Survey Shows Commuters Statewide What isn’t so obvious is the fact that today there are life That took a lot of teamwork, including some science and/or support companies in 85 of North fabulous support from human resources and Carolina’s 100 counties. Also, even if your county other key people at 58 companies across the state, doesn’t yet have one of these companies, people from mostly life science firms. your county are likely to be working for one. Some of them have a hefty commute, to be sure. -

Qualifying Therapeutic Discovery Project Grants
Qualifying Therapeutic Discovery Project Grants The listings below represent the applicants that have been awarded a Qualifying Therapeutic Discovery Program Grant. The project descriptions below were extracted from the applicant certification forms as submitted. Alabama $3,708,304.65 Grants Grants Applicant Name Project Name Awarded for Awarded for 2009 2010 Molecular Heparan Sulfate Agenta Biotechnologies Delivery in Barrier Membranes $59,614.76 $184,864.48 Inc and Soft Tissue Wound Closure DELIVERY OF GROWTH- AGENTA FACTOR CO-RECEPTOR BIOTECHNOLOGIES HEPARAN SULFATE IN $169,808.39 $6,666.50 INC BONE GRAFT SUBSTITUTES FOR REPAIR BioCryst BCX4208 for Treatment of $244,479.25 Pharmaceuticals Inc Gout BioCryst Peramivir for Treatment of $244,479.25 Pharmaceuticals, Inc Influenza BioCryst Forodesine for Treatment of $244,479.25 Pharmaceuticals, Inc CLL and CTCL BioCryst BCX4161 for Treatment of $86,380.89 Pharmaceuticals, Inc. Hereditary Angioedema JAK Inhibitors for Treatment BioCryst of Psoriasis, Ankylosing $244,479.25 Pharmceuticals, Inc. Spondylitis, & Multiple Sclerosis Disease Diagnostic Panel Diatherix Laboratories, Development Utilizing the $244,479.25 Inc. TEM-PCR Method Grants Grants Applicant Name Project Name Awarded for Awarded for 2009 2010 EGEN-001 in Platinum- EGEN INC $244,479.25 Resistant Ovarian Cancer EGEN, INC. EGEN TheraSilence $244,479.25 Development of an Apparatus iCubate, Inc $244,479.25 for Performing am-PCR Phase 1, Open-label Syudy Evaluting the Safety of PNP Therapeutics Inc $244,479.25 Escalating Doses of Ad/PNP- F-araAMP SER-201 for the treatment of Serina Therapeutics, Inc $244,479.25 colorectal and ovarian cancer Development of an Vaxin Inc. -

Annual Report 2020
ANNUAL REPORT 2020 Caring for People’s Health as a Trusted Partner 2020 A Year like none before Peter Goldschmidt CEO Dear Investors, dear Partners, 2020 was for each of us a demanding year. Like all of you, we at STADA had to deal with new challenges and to adapt to a new situation in our jobs as well as at home with our family. I would like to express my gratitude to all employees worldwide. You have made an extraordinary contribution to our society through your tireless efforts. We can be proud of the strong crisis resilience STADA has shown despite challeng- ing market conditions during the pandemic. The company has proven itself to be a growth leader and go-to-partner in Consumer Healthcare and Generics, with a rapidly expanding Specialty Pharma presence in areas such as Parkinson’s and biosimilars. I’m particularly gratified by how our diverse global team has come through the crisis true to our Purpose of “Caring for People’s Health as a Trusted Partner”. Across the organization, people have pulled together, even if they have been physically distant; this close collaboration across continents truly embodies our value One STADA. Keep Supplying Medicines We care for people’s health as a trusted partner by being able to keep supplying medicines, even during lockdowns. Our 6,000 production staff did an outstanding job in these difficult times. STADA provides a broad portfolio from supplements that enable people to stay healthy and support their immune systems through to essential treatments for diseases in nearly every therapeutic area, such as cancer, heart, central nervous system, gastrointestinal and pain relief. -

Ncbiotech Company Directory
NCBiotech Company Directory Many thanks to our sponsors for making this directory possible July 2012 Alston & Bird • BASF • Bayer CropScience Golden LEAF Biomanufacturing Training and Education Center Cannon Research Center • Fujifilm Diosynth Biotechnologies • GlaxoSmithKline NC Community Colleges BioNetwork • Piedmont Triad Research Park Pozen • PPD • Smith Anderson ncbiotech.org/directory ncbiotech.org/directory 2012CoDirectoryCOVER-final_km-5-14-12.indd 2-3 5/14/12 5:02 PM NCBiotech Company Directory 2012 Growing a healthier world, one harvest at a time. Chairman,Our Board task of Directors is simple, yet monumental. To provide enough food for the world, while John L. Atkins,protecting III, FAIA it at the same time. We believe that with the right combination of innovative President andscience, Chief Executivetenacious Officer problem solving and unshakable passion, we can do it. We will meet E. Norris Tolson Science For A Better Life the needs of today while laying a foundation for a better tomorrow. And in doing so, we Senior Vice President, Science andwill Business not only Development grow a healthier world, we will make sure that abundance endures for us all. Kenneth R. Tindall, Ph.D. Learn more at www.BayerCropScience.us. Senior Vice President, Statewide Operations and Economic Development Michael Wilkins REGIONAL OFFICES Vice President, Statewide Operations Steven Casey, MBA Executive Director, Eastern Office Mark Phillips Executive Director, Greater Charlotte Office Cover photos (back and front cover, left to right): NC Research Campus, NCBiotech Center, BASF SE, Executive Director, Piedmont Triad Office NCBiotech Center, UNCW/MARBIONC/Jamie Moncrief, RTI International, East Carolina University, NC Nancy Johnston State University Photo (above) NCBiotech Center Executive Director, Southeastern Office Randall Johnson, MBA NCBiotech Company Directory is produced by Red Hand Media LLC Executive Director, Western Office 5605 77 Center Drive, Suite 101, Charlotte, NC 28217. -
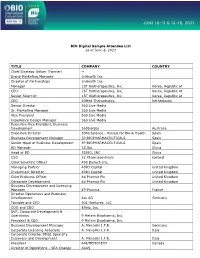
BIO Digital Sample Attendee List As of June 6, 2021 TITLE COMPANY
BIO Digital Sample Attendee List as of June 6, 2021 TITLE COMPANY COUNTRY Chief Strategy Officer (Former) -- Brand Marketing Manager 1nHealth Inc Director of Partnerships 1nHealth Inc Manager 1ST Biotherapeutics, Inc. Korea, Republic of CEO 1ST Biotherapeutics, Inc. Korea, Republic of Senior Scientist 1ST Biotherapeutics, Inc. Korea, Republic of CEO 20Med Therapeutics Netherlands Senior Director 360 Live Media Sr. Marketing Manager 360 Live Media Vice President 360 Live Media Experience Design Manager 360 Live Media Executive Vice President, Business Development 360biolabs Australia Executive Director 3DforScience - Visuals for Bio & Health Spain Business Development Manager 3P BIOPHARMACEUTICALS Spain Senior Head of Business Development 3P BIOPHARMACEUTICALS Spain BD Manager 3S Bio China Head of BD 3SBIO, INC China CSO 3Z Pharmaceuticals Iceland Chief Scientific Officer 490 Biotech Inc. Managing Partner 4BIO Capital United Kingdom Investment Director 4BIO Capital United Kingdom Chief Business Officer 4d Pharma Plc United Kingdom Corporate Development 4d Pharma Plc United Kingdom Business Development and Licensing Manager 4P-Pharma France Director Operations and Business Development 4sc AG Germany Founder and CEO 501 Ventures, LLC COO and CBO 89bio, Inc. SVP, Corporate Development & Operations 9 Meters Biopharma, Inc. President & CEO 9 Meters Biopharma, Inc. Business Development Manager A. Menarini I.F.R. Germany Corporate Licensing Associate A. Menarini I.F.R. Italy Corporate Director BD&L Specialty Discovery and Development A. Menarini I.F.R. Italy President A4I/MEMOTEXT Canada Director of Operations - SEA Change AAAS Founder & CEO Aagami, Inc. Co-Founder and CEO A-Alpha Bio CEO AAVogen CSO Abalone Bio CEO Abalone Bio Inc Chairman Abarceo Pharma AB Sweden CEO Abarceo Pharma AB Sweden CEO Abartys Health, LLC CBO Abbisko Therapeutics Co. -

Immunogenicity and Safety of Fractional Doses of Yellow
medRxiv preprint doi: https://doi.org/10.1101/2020.08.18.20177527; this version posted August 21, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. It is made available under a CC-BY-NC-ND 4.0 International license . 1 Immunogenicity and safety of fractional doses of yellow fever vaccines: a randomized, 2 double-blind, non-inferiority trial 3 4 Aitana Juan-Giner1,*, Derick Kimathi2,3*, Kyra H. Grantz4,5,6, Mainga M. Hamaluba2, Patrick 5 Kazooba7, Patricia Njuguna2, Gamou Fall8, Moussa Dia8, Ndeye S. Bob8, Thomas P. 6 Monath9, Alan D. Barrett10, Joachim Hombach11, Edgar M. Mulogo12, Immaculate Ampeire13, 2 7 7 4,5 7 Henry K. Karanja , Dan Nyehangane , Juliet Mwanga-Amumpaire , Derek A.T. Cummings , 8 Philip Bejon2,3, George M. Warimwe2,3,#, Rebecca F. Grais1,#. 9 10 1 Epicentre, Paris, France 11 2 Kenya Medical Research Institute – Wellcome Trust Research Programme, Kilifi, Kenya 12 3 Centre for Tropical Medicine & Global Health, University of Oxford, UK 13 4 Department of Biology, University of Florida 14 5 Emerging Pathogens Institute, University of Florida 15 6 Department of Epidemiology, Johns Hopkins Bloomberg School of Public Health 16 7 Epicentre Mbarara Research Centre, Uganda 17 8 Institut Pasteur Dakar, Senegal 18 9 Crozet BioPharma LLC, Devens MA, USA 19 10 Sealy Institute for Vaccines Sciences and Department of Pathology, University of Texas 20 Medical Branch, USA 21 11 Immunization, Vaccines & Biologicals,