Bachman's Sparrow (Peucaea Aestivalis)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Comprehensive Multilocus Assessment of Sparrow (Aves: Passerellidae) Relationships ⇑ John Klicka A, , F
Molecular Phylogenetics and Evolution 77 (2014) 177–182 Contents lists available at ScienceDirect Molecular Phylogenetics and Evolution journal homepage: www.elsevier.com/locate/ympev Short Communication A comprehensive multilocus assessment of sparrow (Aves: Passerellidae) relationships ⇑ John Klicka a, , F. Keith Barker b,c, Kevin J. Burns d, Scott M. Lanyon b, Irby J. Lovette e, Jaime A. Chaves f,g, Robert W. Bryson Jr. a a Department of Biology and Burke Museum of Natural History and Culture, University of Washington, Box 353010, Seattle, WA 98195-3010, USA b Department of Ecology, Evolution, and Behavior, University of Minnesota, 100 Ecology Building, 1987 Upper Buford Circle, St. Paul, MN 55108, USA c Bell Museum of Natural History, University of Minnesota, 100 Ecology Building, 1987 Upper Buford Circle, St. Paul, MN 55108, USA d Department of Biology, San Diego State University, San Diego, CA 92182, USA e Fuller Evolutionary Biology Program, Cornell Lab of Ornithology, Cornell University, 159 Sapsucker Woods Road, Ithaca, NY 14950, USA f Department of Biology, University of Miami, 1301 Memorial Drive, Coral Gables, FL 33146, USA g Universidad San Francisco de Quito, USFQ, Colegio de Ciencias Biológicas y Ambientales, y Extensión Galápagos, Campus Cumbayá, Casilla Postal 17-1200-841, Quito, Ecuador article info abstract Article history: The New World sparrows (Emberizidae) are among the best known of songbird groups and have long- Received 6 November 2013 been recognized as one of the prominent components of the New World nine-primaried oscine assem- Revised 16 April 2014 blage. Despite receiving much attention from taxonomists over the years, and only recently using molec- Accepted 21 April 2014 ular methods, was a ‘‘core’’ sparrow clade established allowing the reconstruction of a phylogenetic Available online 30 April 2014 hypothesis that includes the full sampling of sparrow species diversity. -

Sonoran Joint Venture Bird Conservation Plan Version 1.0
Sonoran Joint Venture Bird Conservation Plan Version 1.0 Sonoran Joint Venture 738 N. 5th Avenue, Suite 102 Tucson, AZ 85705 520-882-0047 (phone) 520-882-0037 (fax) www.sonoranjv.org May 2006 Sonoran Joint Venture Bird Conservation Plan Version 1.0 ____________________________________________________________________________________________ Acknowledgments We would like to thank all of the members of the Sonoran Joint Venture Technical Committee for their steadfast work at meetings and for reviews of this document. The following Technical Committee meetings were devoted in part or total to working on the Bird Conservation Plan: Tucson, June 11-12, 2004; Guaymas, October 19-20, 2004; Tucson, January 26-27, 2005; El Palmito, June 2-3, 2005, and Tucson, October 27-29, 2005. Another major contribution to the planning process was the completion of the first round of the northwest Mexico Species Assessment Process on May 10-14, 2004. Without the data contributed and generated by those participants we would not have been able to successfully assess and prioritize all bird species in the SJV area. Writing the Conservation Plan was truly a group effort of many people representing a variety of agencies, NGOs, and universities. Primary contributors are recognized at the beginning of each regional chapter in which they participated. The following agencies and organizations were involved in the plan: Arizona Game and Fish Department, Audubon Arizona, Centro de Investigación Cientifica y de Educación Superior de Ensenada (CICESE), Centro de Investigación de Alimentación y Desarrollo (CIAD), Comisión Nacional de Áreas Naturales Protegidas (CONANP), Instituto del Medio Ambiente y el Desarrollo (IMADES), PRBO Conservation Science, Pronatura Noroeste, Proyecto Corredor Colibrí, Secretaría de Medio Ambiente y Recursos Naturales (SEMARNAT), Sonoran Institute, The Hummingbird Monitoring Network, Tucson Audubon Society, U.S. -

Updating the Distribution of the Sierra Madre Sparrow Xenospiza Baileyi Across Central Mexico: Historical Records, New Localities, and Conservation Perspectives
VOLUME 15, ISSUE 1, ARTICLE 15 Ortega-Álvarez, R., R. Calderón-Parra, U. Martínez Molina, F. Martínez Molina, G. Martínez Molina, Y. Martínez Molina, A. Martínez Villagrán, J. Martínez Freire, R. Vásquez Robles, D. García Loaeza, J. Martínez García, S. García Loaeza, N. I. Garduño López, and L. A. Sánchez-González. 2020. Updating the distribution of the Sierra Madre Sparrow Xenospiza baileyi across central Mexico: historical records, new localities, and conservation perspectives. Avian Conservation and Ecology 15(1):15. https://doi.org/10.5751/ACE-01573-150115 Copyright © 2020 by the author(s). Published here under license by the Resilience Alliance. Research Paper Updating the distribution of the Sierra Madre Sparrow Xenospiza baileyi across central Mexico: historical records, new localities, and conservation perspectives Rubén Ortega-Álvarez 1, Rafael Calderón-Parra 2, Ulises Martínez Molina 2, Fredy Martínez Molina 2, Gabriel Martínez Molina 2, Yuridia Martínez Molina 2, Agustín Martínez Villagrán 2, Josué Martínez Freire 2, Rocío Vásquez Robles 2, Delfino García Loaeza 2, Jaciel Martínez García 2, Sarai García Loaeza 2, Nancy Isabel Garduño López 2 and Luis A. Sánchez-González 1 1Museo de Zoología "Alfonso L. Herrera," Departamento de Biología Evolutiva, Facultad de Ciencias, Universidad Nacional Autónoma de México, Ciudad de México, México, 2Brigada de Monitoreo Biológico Milpa Alta, San Pablo Oztotepec, Milpa Alta, Ciudad de México, México ABSTRACT. The Sierra Madre Sparrow (Xenospiza baileyi) is an endangered species microendemic to subalpine grasslands of central and northwestern Mexico. Given that land use change is highly dynamic in central Mexico, there is a need for a continuous monitoring of the distribution and conservation status of the species across the region. -
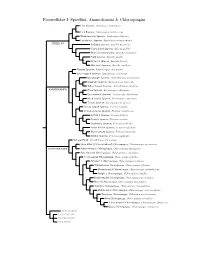
Passerellidae Species Tree
Passerellidae I: Spizellini, Ammodramini & Chlorospingini Lark Sparrow, Chondestes grammacus Lark Bunting, Calamospiza melanocorys Black-throated Sparrow, Amphispiza bilineata Five-striped Sparrow, Amphispiza quinquestriata SPIZELLINI Chipping Sparrow, Spizella passerina Clay-colored Sparrow, Spizella pallida Black-chinned Sparrow, Spizella atrogularis Field Sparrow, Spizella pusilla Brewer’s Sparrow, Spizella breweri Worthen’s Sparrow, Spizella wortheni Tumbes Sparrow, Rhynchospiza stolzmanni Stripe-capped Sparrow, Rhynchospiza strigiceps Grasshopper Sparrow, Ammodramus savannarum Grassland Sparrow, Ammodramus humeralis Yellow-browed Sparrow, Ammodramus aurifrons AMMODRAMINI Olive Sparrow, Arremonops rufivirgatus Green-backed Sparrow, Arremonops chloronotus Black-striped Sparrow, Arremonops conirostris Tocuyo Sparrow, Arremonops tocuyensis Rufous-winged Sparrow, Peucaea carpalis Cinnamon-tailed Sparrow, Peucaea sumichrasti Botteri’s Sparrow, Peucaea botterii Cassin’s Sparrow, Peucaea cassinii Bachman’s Sparrow, Peucaea aestivalis Stripe-headed Sparrow, Peucaea ruficauda Black-chested Sparrow, Peucaea humeralis Bridled Sparrow, Peucaea mystacalis Tanager Finch, Oreothraupis arremonops Short-billed (Yellow-whiskered) Chlorospingus, Chlorospingus parvirostris CHLOROSPINGINI Yellow-throated Chlorospingus, Chlorospingus flavigularis Ashy-throated Chlorospingus, Chlorospingus canigularis Sooty-capped Chlorospingus, Chlorospingus pileatus Wetmore’s Chlorospingus, Chlorospingus wetmorei White-fronted Chlorospingus, Chlorospingus albifrons Brown-headed -
Adaptive Genetic Diversity and Evidence of Population Genetic Structure in the Endangered Sierra Madre Sparrow (Xenospiza Baileyi)
PLOS ONE RESEARCH ARTICLE Adaptive genetic diversity and evidence of population genetic structure in the endangered Sierra Madre Sparrow (Xenospiza baileyi) 1 1 2 Jose G. Ham-DueñasID , Ricardo Canales-del-CastilloID *, Gary Voelker , Irene Ruvalcaba-Ortega1, Carlos E. Aguirre-Caldero n3, Jose I. GonzaÂlez-Rojas1 a1111111111 1 Laboratorio de BiologõÂa de la ConservacioÂn y Desarrollo Sustentable. Cd. Universitaria, Universidad AutoÂnoma de Nuevo LeoÂn, Facultad de Ciencias BioloÂgicas, San NicolaÂs de los Garza, Nuevo LeoÂn, MeÂxico, a1111111111 2 Department of Wildlife and Fisheries Sciences, Biodiversity Research and Teaching Collections, Texas a1111111111 A&M University, College Station, Texas, United States of America, 3 Instituto TecnoloÂgico de El Salto, El a1111111111 Salto, P.N., Durango, Mexico a1111111111 * [email protected] Abstract OPEN ACCESS Citation: Ham-Dueñas JG, Canales-del-Castillo R, The magnitude and distribution of genetic diversity through space and time can provide Voelker G, Ruvalcaba-Ortega I, Aguirre-CalderoÂn useful information relating to evolutionary potential and conservation status in threatened CE, GonzaÂlez-Rojas JI (2020) Adaptive genetic species. In assessing genetic diversity in species that are of conservation concern, sev- diversity and evidence of population genetic eral studies have focused on the use of Toll-like receptors (TLRs). TLRs are innate structure in the endangered Sierra Madre Sparrow (Xenospiza baileyi). PLoS ONE 15(4): e0232282. immune genes related to pathogen resistance, and polymorphisms may reflect not only https://doi.org/10.1371/journal.pone.0232282 levels of functional diversity, but may also be used to assess genetic diversity within and Editor: Arnar Palsson, University of Iceland, among populations. -

NEWS and NOTES by Paul Hess
NEWS AND NOTES by Paul Hess “Lilian’s” Meadowlark für Ornithologie 135:28), but no details about the findings have been published. Ornithologist Harry C. Oberholser was intrigued by mead - At last, a large-scale genetic report by F. Keith Barker, Ar - owlarks collected in 1929 near Arizona’s Huachuca Moun - ion J. Vandergon, and Scott M. Lanyon arrived in 2008 tains, because their size, structure, and plumage differed (Auk 125:869–879). They examined two mitochondrial from those of the Western Meadowlark and from known DNA (mtDNA) genes and one nuclear locus in 14 Eastern variations of Eastern Meadowlark. In 1930 he described the Meadowlark subspecies throughout the North American, specimens as a new Eastern Meadowlark subspecies and Central American, and South American range. named it lilianae to honor Lilian Baldwin, who had donat - The results indicate a long history of evolutionary diver - ed the collection ( Scientific Publications of the Cleveland Mu - gence between lilianae and all except one other Eastern seum of Natural History 1:83–124). Meadowlark subspecies, auropectoralis of central and We now know it as “Lilian’s” Meadowlark, of desert grasslands in the southwestern U.S. and northern Mexico, where it is distinguishable from Western Meadowlark with careful study by eye and ear. In identification guides, lilianae re - ceived little attention and no illustration until the National Geographic Field Guide to the Birds of North America in 1983. Kevin J. Zimmer of - fered tips in Birding (August 1984, pp. 155–156) and in his books The Western Bird Watcher (Prentice-Hall 1985) and Birding in the American West (Cornell University Press 2000). -

The Current Status of Sinaloa Martin Progne Sinaloae
Cotinga 32 The current status of Sinaloa Martin Progne sinaloae Nicholas A. Lethaby and Jon R. King Received 24 February 2009; final revision accepted 13 November 2009 first published online 16 March 2010 Cotinga 32 (2010): 90–95 La Golondrina de Sinaloa Progne sinaloae es una especie categorizada como Datos Deficientes y endémica reproductiva de la Sierra Madre Occidental y las montañas Volcánicas Centrales de México. Sus áreas de invernada son desconocidas. A pesar de que su estado es descrito como poco común a relativamente común, no hay localidades donde la especie sea vista regularmente o en números significativos. Históricamente, los registros han sido de colonias reproductivas de 15–30 aves o más; pero los registros acumulados por los autores a lo largo de las últimas dos décadas han sido en su mayoría de migrantes en las tierras bajas, sin registros reproductivos definitivos excepto por una colonia recientemente descubierta de solo 2–6 individuos en Sinaloa. La Golondrina de Sinaloa es inherentemente conspicua y los machos son fáciles de identificar, por lo que es una especie difícil de pasar desapercibida. Parece ser poco común y altamente local en el mejor de los casos, y posiblemente muy rara. Monitoreos formales deberían ser desarrollados para evaluar con precisión el actual estado de conservación de la especie y sus tendencias poblacionales. The Sierra Madre Occidental is an extensive There are potentially several reasons why mountain range in north-west Mexico. Although the apparent rarity of Sinaloa Martin has gone much of the sierra appears to be extremely remote, unnoticed. subsistence farming, ranching, hunting and logging 1. -

Florida Grasshopper Sparrow (Ammodramus Savannarum Floridanus) Conspecific Attraction Experiment
FLORIDA GRASSHOPPER SPARROW (AMMODRAMUS SAVANNARUM FLORIDANUS) CONSPECIFIC ATTRACTION EXPERIMENT THOMAS VIRZI ECOSTUDIES INSTITUTE P.O. BOX 735 EAST OLYMPIA, WA 98540 [email protected] REPORT TO THE U.S. FISH & WILDLIFE SERVICE (SOUTH FLORIDA ECOLOGICAL SERVICES, VERO BEACH, FL) AND THE FISH & WILDLIFE FOUNDATION OF FLORIDA (TALLAHASSEE, FL) DECEMBER 2015 Contents 1.0 EXECUTIVE SUMMARY .............................................................................................................................. 3 2.0 INTRODUCTION ......................................................................................................................................... 7 2.1 PURPOSE ......................................................................................................................................................... 7 2.2 FIGURES ........................................................................................................................................................ 11 3.0 PLAYBACK EXPERIMENT .......................................................................................................................... 13 3.1 BACKGROUND ................................................................................................................................................ 13 3.2 METHODS ..................................................................................................................................................... 13 3.3 RESULTS AND DISCUSSION ............................................................................................................................... -

Proposals 2018-C
AOS Classification Committee – North and Middle America Proposal Set 2018-C 1 March 2018 No. Page Title 01 02 Adopt (a) a revised linear sequence and (b) a subfamily classification for the Accipitridae 02 10 Split Yellow Warbler (Setophaga petechia) into two species 03 25 Revise the classification and linear sequence of the Tyrannoidea (with amendment) 04 39 Split Cory's Shearwater (Calonectris diomedea) into two species 05 42 Split Puffinus boydi from Audubon’s Shearwater P. lherminieri 06 48 (a) Split extralimital Gracula indica from Hill Myna G. religiosa and (b) move G. religiosa from the main list to Appendix 1 07 51 Split Melozone occipitalis from White-eared Ground-Sparrow M. leucotis 08 61 Split White-collared Seedeater (Sporophila torqueola) into two species (with amendment) 09 72 Lump Taiga Bean-Goose Anser fabalis and Tundra Bean-Goose A. serrirostris 10 78 Recognize Mexican Duck Anas diazi as a species 11 87 Transfer Loxigilla portoricensis and L. violacea to Melopyrrha 12 90 Split Gray Nightjar Caprimulgus indicus into three species, recognizing (a) C. jotaka and (b) C. phalaena 13 93 Split Barn Owl (Tyto alba) into three species 14 99 Split LeConte’s Thrasher (Toxostoma lecontei) into two species 15 105 Revise generic assignments of New World “grassland” sparrows 1 2018-C-1 N&MA Classification Committee pp. 87-105 Adopt (a) a revised linear sequence and (b) a subfamily classification for the Accipitridae Background: Our current linear sequence of the Accipitridae, which places all the kites at the beginning, followed by the harpy and sea eagles, accipiters and harriers, buteonines, and finally the booted eagles, follows the revised Peters classification of the group (Stresemann and Amadon 1979). -

Behavioral, Morphological, and Ecological Trait Evolution in Two Clades of New World Sparrows (Aimophila and Peucaea, Passerellidae)
Behavioral, morphological, and ecological trait evolution in two clades of New World Sparrows (Aimophila and Peucaea, Passerellidae) Carla Cicero1, Nicholas A. Mason1,4, Lauryn Benedict2 and James D. Rising3,† 1 Museum of Vertebrate Zoology, University of California, Berkeley, Berkeley, CA, United States of America 2 School of Biological Sciences, University of Northern Colorado, Greeley, CO, United States of America 3 Department of Ecology and Evolutionary Biology, University of Toronto, Toronto, Ontario, Canada 4 Current affiliation: Museum of Natural Science and Department of Biological Sciences, Louisiana State University, Baton Rouge, Louisiana, United States of America † Deceased. ABSTRACT The New World sparrows (Passerellidae) are a large, diverse group of songbirds that vary in morphology, behavior, and ecology. Thus, they are excellent for studying trait evolution in a phylogenetic framework. We examined lability versus conservatism in morphological and behavioral traits in two related clades of sparrows (Aimophila, Peucaea), and assessed whether habitat has played an important role in trait evolution. We first inferred a multi-locus phylogeny which we used to reconstruct ancestral states, and then quantified phylogenetic signal among morphological and behavioral traits in these clades and in New World sparrows more broadly. Behavioral traits have a stronger phylogenetic signal than morphological traits. Specifically, vocal duets and song structure are the most highly conserved traits, and nesting behavior appears to be maintained within clades. Furthermore, we found a strong correlation between open habitat and unpatterned plumage, complex song, and ground nesting. However, even within lineages that share the same habitat type, species vary in nesting, plumage pattern, song complexity, and duetting. Our findings highlight trade-offs between behavior, morphology, and ecology in sparrow diversification. -

Species Relationships in the Avian Genus Aimophila
SPECIES RELATIONSHIPS IN THE AVIAN GENUS dIAdOPI-llLd BY LARRY L. WOLF Museumof VertebrateZoology Universityof California ORNITHOLOGICAL MONOGRAPHS NO. 23 PUBLISHED BY THE AMERICAN ORNITHOLOGISTS' UNION 1977 SPECIES RELATIONSHIPS IN THE AVIAN GENUS •IZA4tOPZ--ZZL•I ORNITHOLOGICAL MONOGRAPHS This series,published by the American Ornithologists'Union, has been establishedfor major papers too long for inclusionin the Union's journal, The Auk. Publicationhas been made possiblethrough the generosityof Mrs. Carll Tucker and the Marcia Brady Tucker Foundation,Inc. Correspondenceconcerning manuscripts for publicationin the seriesshould be addressedto the Editor, Dr. John William Hardy, Departmentof Natural Science,The Florida StateMuseum, University of Florida, Gainesville,Florida 32611. Copiesof OrnithologicalMonographs may be orderedfrom the Assistant to the Treasurerof the AOU, Glen E. Woolfender•Department of Biology, Universityof SouthFlorida, Tampa, Florida 33620. (See price list on back and inside back cover.) OrnithologicalMonographs No. 23, viii + 220 pp. Editor of A.O.U. Monographs,John William Hardy SpecialAssociate Editors of this issue,John P. Hubbard, Dela- ware Museum of Natural History, Greenville,Delaware 19807, and Ralph J. Raitt, Departmentof Biology,New Mexico State University,Las Cruces,New Mexico 88001 AssistantEditor, June B. Gabaldon Author, Larry L. Wolf, Departmentof Biology, SyracuseUni- versity, Syracuse,New York 13210 First received, 24 January 1974; accepted,2 February 1976; final revisioncompleted, 9 January 1976 Issued February 23, 1977 Price (includeslong-play phono-discalbum) $12.00 prepaid ($10.50 to AOU Members) Library of CongressCatalogue Card Number 77-73658 Primedby the Allen Press,Inc., Lawrence,Kansas 66044 Copyright ¸ by American Ornithologists'Union, 1977 ii SPECIES RELATIONSHIPS IN THE AVIAN GENUS •1IMOPHIL•I BY LARRY L. -

On the Sierra Madre Sparrow, Xenospiza Baileyi, of Mexico
ON THE SIERRA MADRE SPARROW, XENOSPIZA BAILEYI, OF MEXICO ROBERTW. DICKERMAN,ALLAN R. PHILLIPS, AND DWAIN W. WARNER THE SierraMadre or Bailey'sSparrow, Xenospiza baileyi, was described as a new genus and speciesfrom the high mountainsof Durango and Jaliscoless than 35 yearsago (O. Bangs,Proc. •VewEngland Zool. Cl., 12: 85-88, 1931). It has sincebeen consideredrare, and even 19 years ago, when F. A. Pitelka (Condor, 49: 199-203, 1947) describeda new subspecies,only 11 specimenswere known, of whichhe had at handonly 1 from La Cima, Distrito Federal, and 3 from westernMexico. Today the populationfrom the sacatonbunch grass areas of La Cima, in the pass betweenthe Valleyof Mexicoand Cuernavaca,is well representedby series in the collectionsof Phillips and the University of Minnesota Museum of Natural History. These seriesinclude specimens in all the plumage stagesfrom stubby-tailedjuveniles to worn adults and those having recently completedthe prebasicmolt. Other specimensare scattered through several museumsin the United States. The populationof Du- rangois now representedby additionalspecimens in the collectionof the Robert T. Moore Laboratoryof Zoology,Occidental College. With the accumulationof this material,and notesfrom severalseasons' field work by the authors,it is a propitioustime to gather togetherthe information on this restricted endemic Mexican sparrow. ACI•N'OWLEDGEM. Ei•TS We wish to expressour appreciation to the curators of the following collections for permitting us to examine materials in their care: American Museum of Natural History, New York; Denver Museum of Natural History; Museum of Comparative Zoology, Harvard College, Cambridge, Massachusetts; Robert T. Moore Collection, Occidental College, Los Angeles, California; and Museum of Vertebrate Zoology, University of California, Berkeley, California.