An Interstitial Deletion-Insertion Involving Chromosomes 2P25.3 and Xq27.1, Near SOX3, Causes X-Linked Recessive Hypoparathyroidism
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Identification of Potential Proteases for Abdominal Aortic Aneurysm by Weighted Gene Coexpression Network Analysis
Genome Identification of potential proteases for abdominal aortic aneurysm by weighted gene coexpression network analysis Journal: Genome Manuscript ID gen-2020-0041.R1 Manuscript Type: Article Date Submitted by the 28-Jun-2020 Author: Complete List of Authors: Zhang, Hui; Peking Union Medical College Hospital, Department of Vascular Surgery Yang, Dan; Chinese Academy of Medical Sciences and Peking Union Medical College, Department of Computational Biology and Bioinformatics,Draft Institute of Medicinal Plant Development Chen, Siliang; Peking Union Medical College Hospital, Department of Vascular Surgery Li, Fangda; Peking Union Medical College Hospital, Department of Vascular Surgery Cui, Liqiang; Peking Union Medical College Hospital, Department of Vascular Surgery Liu, Zhili; Peking Union Medical College Hospital, Department of Vascular Surgery Shao, Jiang; Peking Union Medical College Hospital, Department of Vascular Surgery Chen, Yuexin; Peking Union Medical College Hospital, Department of Vascular Surgery Liu, Bao; Peking Union Medical College Hospital, Department of Vascular Surgery Zheng, Yuehong; Peking Union Medical College Hospital, Department of Vascular Surgery Abdominal aortic aneurysm, next-generation sequencing, WGCNA, Keyword: proteases, matrix metalloproteinase Is the invited manuscript for consideration in a Special Not applicable (regular submission) Issue? : https://mc06.manuscriptcentral.com/genome-pubs Page 1 of 35 Genome 1 Identification of potential proteases for abdominal aortic aneurysm by weighted gene 2 coexpression network analysis 3 Short title: WGCNA identifies crucial proteases in AAA 4 5 Hui Zhang1, Dan Yang2, Siliang Chen1, Fangda Li1, Liqiang Cui1, Zhili Liu1, Jiang Shao1, Yuexin 6 Chen1, Bao Liu1, Yuehong Zheng1. 7 1Department of Vascular Surgery, Peking Union Medical College Hospital, Beijing 100730, PR 8 China; 2Department of Computational Biology and Bioinformatics, Institute of Medicinal Plant 9 Development, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 10 100730, PR China. -

Investigation of Candidate Genes and Mechanisms Underlying Obesity
Prashanth et al. BMC Endocrine Disorders (2021) 21:80 https://doi.org/10.1186/s12902-021-00718-5 RESEARCH ARTICLE Open Access Investigation of candidate genes and mechanisms underlying obesity associated type 2 diabetes mellitus using bioinformatics analysis and screening of small drug molecules G. Prashanth1 , Basavaraj Vastrad2 , Anandkumar Tengli3 , Chanabasayya Vastrad4* and Iranna Kotturshetti5 Abstract Background: Obesity associated type 2 diabetes mellitus is a metabolic disorder ; however, the etiology of obesity associated type 2 diabetes mellitus remains largely unknown. There is an urgent need to further broaden the understanding of the molecular mechanism associated in obesity associated type 2 diabetes mellitus. Methods: To screen the differentially expressed genes (DEGs) that might play essential roles in obesity associated type 2 diabetes mellitus, the publicly available expression profiling by high throughput sequencing data (GSE143319) was downloaded and screened for DEGs. Then, Gene Ontology (GO) and REACTOME pathway enrichment analysis were performed. The protein - protein interaction network, miRNA - target genes regulatory network and TF-target gene regulatory network were constructed and analyzed for identification of hub and target genes. The hub genes were validated by receiver operating characteristic (ROC) curve analysis and RT- PCR analysis. Finally, a molecular docking study was performed on over expressed proteins to predict the target small drug molecules. Results: A total of 820 DEGs were identified between -

SUPPLEMENTARY MATERIAL Human Engineered Skeletal Muscle
SUPPLEMENTARY MATERIAL Human engineered skeletal muscle of hypaxial origin from pluripotent stem cells with advanced function and regenerative capacity Mina Shahriyari1,2, Md Rezaul Islam3, M. Sadman Sakib3, Anastasia Rika1,2, Dennis Krüger3, Lalit Kaurani3, Harithaa Anandakumar1,2, Orr Shomroni4, Matthias Schmidt5, Jana Zschüntzsch5, Jens Schmidt5, Gabriela Salinas-Riester4, Andreas Unger6, Wolfgang A. Linke6, André Fischer3,7, Wolfram-Hubertus Zimmermann1,2,3,7,8*, Malte Tiburcy1,2* 1 Institute of Pharmacology and Toxicology, University Medical Center Göttingen, Göttingen, Germany. 2 DZHK (German Center for Cardiovascular Research), partner site Göttingen. 3 Department for Epigenetics and Systems Medicine in Neurodegenerative Diseases, German Center for Neurodegenerative Diseases (DZNE) Göttingen, Göttingen, Germany 4 NGS Integrative Genomics Core Unit, Institute of Human Genetics, University Medical Center Göttingen, Göttingen, Germany 5 Department of Neurology, University Medical Center Göttingen, Göttingen, Germany 6 Institute of Physiology II, University of Münster, D-48149 Münster, Germany 7 Cluster of Excellence "Multiscale Bioimaging: from Molecular Machines to Networks of Excitable Cells" (MBExC), University of Göttingen, Germany; 8 Fraunhofer Institute for Translational Medicine and Pharmacology (ITMP), Göttingen, Germany Supplementary table 1 related to Figure 2: Biological process annotation of coexpression clusters. Cluster Biological process annotated Enriched GO terms identifier Black Migrating limb progenitors Muscle -

© Ferrata Storti Foundation
Red Cell Biology & its Disorders ARTICLE ATP11C is a major flippase in human erythrocytes and its defect causes congenital Ferrata Storti EUROPEAN HEMATOLOGY Foundation hemolytic anemia ASSOCIATION Nobuto Arashiki,1 Yuichi Takakuwa,1 Narla Mohandas,2 John Hale,2 Kenichi Yoshida,3 Hiromi Ogura,4 Taiju Utsugisawa,4 Shouichi Ohga,5 Satoru Miyano,6 Seishi Ogawa,3 Seiji Kojima,7 and Hitoshi Kanno,4,8 1Department of Biochemistry, School of Medicine, Tokyo Women’s Medical University, Japan; 2Red Cell Physiology Laboratory, New York Blood Center, NY, USA; 3Department of Pathology and Tumor Biology, Graduate School of Medicine, Kyoto University, Japan; 4Department of Transfusion Medicine and Cell Processing, School of Medicine, Tokyo Women’s Medical University, Japan; 5Department of Pediatrics, Graduate School of Medicine, Yamaguchi University, Japan; 6Laboratory of DNA Information Analysis, Human Genome Center, Institute of Medical Science, The University of Tokyo, Japan; 7Department of Pediatrics, Graduate School of Medicine, Nagoya University, Japan; and 8Division of Genomic Medicine, Department of Advanced Biomedical Engineering and Haematologica 2016 Science, Graduate School of Medicine, Tokyo Women's Medical University, Japan Volume 101(5):559-565 ABSTRACT hosphatidylserine is localized exclusively to the inner leaflet of the membrane lipid bilayer of most cells, including erythrocytes. This Pasymmetric distribution is critical for the survival of erythrocytes in circulation since externalized phosphatidylserine is a phagocytic signal for splenic macrophages. Flippases are P-IV ATPase family proteins that active- ly transport phosphatidylserine from the outer to inner leaflet. It has not yet been determined which of the 14 members of this family of proteins is the flippase in human erythrocytes. -

Human Induced Pluripotent Stem Cell–Derived Podocytes Mature Into Vascularized Glomeruli Upon Experimental Transplantation
BASIC RESEARCH www.jasn.org Human Induced Pluripotent Stem Cell–Derived Podocytes Mature into Vascularized Glomeruli upon Experimental Transplantation † Sazia Sharmin,* Atsuhiro Taguchi,* Yusuke Kaku,* Yasuhiro Yoshimura,* Tomoko Ohmori,* ‡ † ‡ Tetsushi Sakuma, Masashi Mukoyama, Takashi Yamamoto, Hidetake Kurihara,§ and | Ryuichi Nishinakamura* *Department of Kidney Development, Institute of Molecular Embryology and Genetics, and †Department of Nephrology, Faculty of Life Sciences, Kumamoto University, Kumamoto, Japan; ‡Department of Mathematical and Life Sciences, Graduate School of Science, Hiroshima University, Hiroshima, Japan; §Division of Anatomy, Juntendo University School of Medicine, Tokyo, Japan; and |Japan Science and Technology Agency, CREST, Kumamoto, Japan ABSTRACT Glomerular podocytes express proteins, such as nephrin, that constitute the slit diaphragm, thereby contributing to the filtration process in the kidney. Glomerular development has been analyzed mainly in mice, whereas analysis of human kidney development has been minimal because of limited access to embryonic kidneys. We previously reported the induction of three-dimensional primordial glomeruli from human induced pluripotent stem (iPS) cells. Here, using transcription activator–like effector nuclease-mediated homologous recombination, we generated human iPS cell lines that express green fluorescent protein (GFP) in the NPHS1 locus, which encodes nephrin, and we show that GFP expression facilitated accurate visualization of nephrin-positive podocyte formation in -

Identification of Genetic Factors Underpinning Phenotypic Heterogeneity in Huntington’S Disease and Other Neurodegenerative Disorders
Identification of genetic factors underpinning phenotypic heterogeneity in Huntington’s disease and other neurodegenerative disorders. By Dr Davina J Hensman Moss A thesis submitted to University College London for the degree of Doctor of Philosophy Department of Neurodegenerative Disease Institute of Neurology University College London (UCL) 2020 1 I, Davina Hensman Moss confirm that the work presented in this thesis is my own. Where information has been derived from other sources, I confirm that this has been indicated in the thesis. Collaborative work is also indicated in this thesis. Signature: Date: 2 Abstract Neurodegenerative diseases including Huntington’s disease (HD), the spinocerebellar ataxias and C9orf72 associated Amyotrophic Lateral Sclerosis / Frontotemporal dementia (ALS/FTD) do not present and progress in the same way in all patients. Instead there is phenotypic variability in age at onset, progression and symptoms. Understanding this variability is not only clinically valuable, but identification of the genetic factors underpinning this variability has the potential to highlight genes and pathways which may be amenable to therapeutic manipulation, hence help find drugs for these devastating and currently incurable diseases. Identification of genetic modifiers of neurodegenerative diseases is the overarching aim of this thesis. To identify genetic variants which modify disease progression it is first necessary to have a detailed characterization of the disease and its trajectory over time. In this thesis clinical data from the TRACK-HD studies, for which I collected data as a clinical fellow, was used to study disease progression over time in HD, and give subjects a progression score for subsequent analysis. In this thesis I show blood transcriptomic signatures of HD status and stage which parallel HD brain and overlap with Alzheimer’s disease brain. -

Epigenetic Mechanisms Are Involved in the Oncogenic Properties of ZNF518B in Colorectal Cancer
Epigenetic mechanisms are involved in the oncogenic properties of ZNF518B in colorectal cancer Francisco Gimeno-Valiente, Ángela L. Riffo-Campos, Luis Torres, Noelia Tarazona, Valentina Gambardella, Andrés Cervantes, Gerardo López-Rodas, Luis Franco and Josefa Castillo SUPPLEMENTARY METHODS 1. Selection of genomic sequences for ChIP analysis To select the sequences for ChIP analysis in the five putative target genes, namely, PADI3, ZDHHC2, RGS4, EFNA5 and KAT2B, the genomic region corresponding to the gene was downloaded from Ensembl. Then, zoom was applied to see in detail the promoter, enhancers and regulatory sequences. The details for HCT116 cells were then recovered and the target sequences for factor binding examined. Obviously, there are not data for ZNF518B, but special attention was paid to the target sequences of other zinc-finger containing factors. Finally, the regions that may putatively bind ZNF518B were selected and primers defining amplicons spanning such sequences were searched out. Supplementary Figure S3 gives the location of the amplicons used in each gene. 2. Obtaining the raw data and generating the BAM files for in silico analysis of the effects of EHMT2 and EZH2 silencing The data of siEZH2 (SRR6384524), siG9a (SRR6384526) and siNon-target (SRR6384521) in HCT116 cell line, were downloaded from SRA (Bioproject PRJNA422822, https://www.ncbi. nlm.nih.gov/bioproject/), using SRA-tolkit (https://ncbi.github.io/sra-tools/). All data correspond to RNAseq single end. doBasics = TRUE doAll = FALSE $ fastq-dump -I --split-files SRR6384524 Data quality was checked using the software fastqc (https://www.bioinformatics.babraham. ac.uk /projects/fastqc/). The first low quality removing nucleotides were removed using FASTX- Toolkit (http://hannonlab.cshl.edu/fastxtoolkit/). -

Supplementary Tables S1-S3
Supplementary Table S1: Real time RT-PCR primers COX-2 Forward 5’- CCACTTCAAGGGAGTCTGGA -3’ Reverse 5’- AAGGGCCCTGGTGTAGTAGG -3’ Wnt5a Forward 5’- TGAATAACCCTGTTCAGATGTCA -3’ Reverse 5’- TGTACTGCATGTGGTCCTGA -3’ Spp1 Forward 5'- GACCCATCTCAGAAGCAGAA -3' Reverse 5'- TTCGTCAGATTCATCCGAGT -3' CUGBP2 Forward 5’- ATGCAACAGCTCAACACTGC -3’ Reverse 5’- CAGCGTTGCCAGATTCTGTA -3’ Supplementary Table S2: Genes synergistically regulated by oncogenic Ras and TGF-β AU-rich probe_id Gene Name Gene Symbol element Fold change RasV12 + TGF-β RasV12 TGF-β 1368519_at serine (or cysteine) peptidase inhibitor, clade E, member 1 Serpine1 ARE 42.22 5.53 75.28 1373000_at sushi-repeat-containing protein, X-linked 2 (predicted) Srpx2 19.24 25.59 73.63 1383486_at Transcribed locus --- ARE 5.93 27.94 52.85 1367581_a_at secreted phosphoprotein 1 Spp1 2.46 19.28 49.76 1368359_a_at VGF nerve growth factor inducible Vgf 3.11 4.61 48.10 1392618_at Transcribed locus --- ARE 3.48 24.30 45.76 1398302_at prolactin-like protein F Prlpf ARE 1.39 3.29 45.23 1392264_s_at serine (or cysteine) peptidase inhibitor, clade E, member 1 Serpine1 ARE 24.92 3.67 40.09 1391022_at laminin, beta 3 Lamb3 2.13 3.31 38.15 1384605_at Transcribed locus --- 2.94 14.57 37.91 1367973_at chemokine (C-C motif) ligand 2 Ccl2 ARE 5.47 17.28 37.90 1369249_at progressive ankylosis homolog (mouse) Ank ARE 3.12 8.33 33.58 1398479_at ryanodine receptor 3 Ryr3 ARE 1.42 9.28 29.65 1371194_at tumor necrosis factor alpha induced protein 6 Tnfaip6 ARE 2.95 7.90 29.24 1386344_at Progressive ankylosis homolog (mouse) -

An Interstitial Deletion-Insertion Involving Chromosomes 2P25.3 and Xq27.1, Near SOX3, Causes X-Linked Recessive Hypoparathyroidism Michael R
Washington University School of Medicine Digital Commons@Becker Open Access Publications 2005 An interstitial deletion-insertion involving chromosomes 2p25.3 and Xq27.1, near SOX3, causes X-linked recessive hypoparathyroidism Michael R. Bowl University of Oxford Andrew Nesbit University of Oxford Brian Harding University of Oxford Elaine Levy University of Oxford Andrew Jefferson University of Oxford See next page for additional authors Follow this and additional works at: https://digitalcommons.wustl.edu/open_access_pubs Recommended Citation Bowl, Michael R.; Nesbit, Andrew; Harding, Brian; Levy, Elaine; Jefferson, Andrew; Volpi, Emanuela; Rizzoti, Karine; Lovell-Badge, Robin; Schlessinger, David; Whyte, Michael P.; and Thakker, Rajesh V., ,"An interstitial deletion-insertion involving chromosomes 2p25.3 and Xq27.1, near SOX3, causes X-linked recessive hypoparathyroidism." The ourJ nal of Clinical Investigation.,. 2822-2831. (2005). https://digitalcommons.wustl.edu/open_access_pubs/1524 This Open Access Publication is brought to you for free and open access by Digital Commons@Becker. It has been accepted for inclusion in Open Access Publications by an authorized administrator of Digital Commons@Becker. For more information, please contact [email protected]. Authors Michael R. Bowl, Andrew Nesbit, Brian Harding, Elaine Levy, Andrew Jefferson, Emanuela Volpi, Karine Rizzoti, Robin Lovell-Badge, David Schlessinger, Michael P. Whyte, and Rajesh V. Thakker This open access publication is available at Digital Commons@Becker: https://digitalcommons.wustl.edu/open_access_pubs/1524 Downloaded on July 12, 2013. The Journal of Clinical Investigation. More information at www.jci.org/articles/view/24156 3FTFBSDIBSUJDMF "OJOUFSTUJUJBMEFMFUJPOJOTFSUJPOJOWPMWJOH DISPNPTPNFTQBOE9R OFBS409 DBVTFT9MJOLFESFDFTTJWFIZQPQBSBUIZSPJEJTN Michael R. Bowl,1 M. Andrew Nesbit,1 Brian Harding,1 Elaine Levy,2 Andrew Jefferson,2 Emanuela Volpi,2 Karine Rizzoti,3 Robin Lovell-Badge,3 David Schlessinger,4 Michael P. -

Autocrine IFN Signaling Inducing Profibrotic Fibroblast Responses By
Downloaded from http://www.jimmunol.org/ by guest on September 23, 2021 Inducing is online at: average * The Journal of Immunology , 11 of which you can access for free at: 2013; 191:2956-2966; Prepublished online 16 from submission to initial decision 4 weeks from acceptance to publication August 2013; doi: 10.4049/jimmunol.1300376 http://www.jimmunol.org/content/191/6/2956 A Synthetic TLR3 Ligand Mitigates Profibrotic Fibroblast Responses by Autocrine IFN Signaling Feng Fang, Kohtaro Ooka, Xiaoyong Sun, Ruchi Shah, Swati Bhattacharyya, Jun Wei and John Varga J Immunol cites 49 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html http://www.jimmunol.org/content/suppl/2013/08/20/jimmunol.130037 6.DC1 This article http://www.jimmunol.org/content/191/6/2956.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2013 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 23, 2021. The Journal of Immunology A Synthetic TLR3 Ligand Mitigates Profibrotic Fibroblast Responses by Inducing Autocrine IFN Signaling Feng Fang,* Kohtaro Ooka,* Xiaoyong Sun,† Ruchi Shah,* Swati Bhattacharyya,* Jun Wei,* and John Varga* Activation of TLR3 by exogenous microbial ligands or endogenous injury-associated ligands leads to production of type I IFN. -
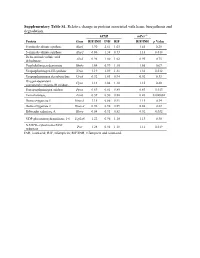
Supplementary Table S1. Relative Change in Proteins Associated with Heme Biosynthesis and Degradation
Supplementary Table S1. Relative change in proteins associated with heme biosynthesis and degradation. hPXR mPxr–/– Protein Gene RIF/INH INH RIF RIF/INH p Value 5-aminolevulinate synthase Alas1 1.90 2.61 1.05 1.41 0.28 5-aminolevulinate synthase Alas2 0.86 1.38 0.73 1.18 0.018 Delta-aminolevulinic acid Alad 0.96 1.00 1.02 0.95 0.75 dehydratase Porphobilinogen deaminase Hmbs 1.04 0.99 1.10 1.05 0.67 Uroporphyrinogen-III synthase Uros 1.19 1.09 1.31 1.38 0.012 Uroporphyrinogen decarboxylase Urod 0.92 1.03 0.94 0.92 0.33 Oxygen-dependent Cpox 1.13 1.04 1.18 1.15 0.20 coproporphyrinogen-III oxidase, Protoporphyrinogen oxidase Ppox 0.69 0.81 0.85 0.83 0.013 Ferrochelatase, Fech 0.39 0.50 0.88 0.43 0.000002 Heme oxygenase 1 Hmox1 1.15 0.86 0.91 1.11 0.34 Heme oxygenase 2 Hmox2 0.96 0.98 0.89 0.88 0.22 Biliverdin reductase A Blvra 0.84 0.92 0.82 0.92 0.032 UDP-glucuronosyltransferase 1-6 Ugt1a6 1.22 0.96 1.10 1.13 0.30 NADPH--cytochrome P450 Por 1.28 0.92 1.18 1.12 0.019 reductase INH, isoniazid; RIF, rifampicin; RIF/INH, rifampicin and isoniazid. Supplementary Table S2. Relative change in protein nuclear receptors. hPXR mPxr–/– Protein Gene RIF/INH INH RIF RIF/INH p Value Aryl hydrocarbon receptor Ahr 1.09 0.91 1.00 1.26 0.092 Hepatocyte nuclear factor Hnf1a 0.87 0.97 0.82 0.79 0.027 1-alpha Hepatocyte nuclear factor Hnf4a 0.95 1.05 0.97 1.08 0.20 4-alpha Oxysterols receptor LXR- Nr1h3 0.94 1.16 1.03 1.02 0.42 alpha Bile acid receptor Nr1h4 1.05 1.17 0.98 1.19 0.12 Retinoic acid receptor Rxra 0.88 1.03 0.83 0.95 0.12 RXR-alpha Peroxisome proliferator-