Chapter 1 Chemistry of Non-Aqueous Solutions
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Kinetic and Equilibrium Acidities of Nitrocycloalkanes J
Kinetic and Equilibrium Acidities of Nitrocycloalkanes J. Org. Chem., Vol. 43, No. 16, 1978 3113 Acknowledgment. We are grateful to the National Science 13) M. Eigen, Angew, Chem., lnt. Ed. Engl., 3, l(1964). 14) E. Grunwald and D. Eustace, ref IO, Chapter 4,have shown that usually Foundation and the National Institutes of Health for support one or two water molecules separate the reactants in proton transfers of of this research. Comments of a referee and of Professor A. J. this type. Kresge were helpful in preparing a revised version of this 15) R. A. More O’Ferrall, ref IO, Chapter 8. 16) R. A. Ogg and M. Polyani, Trans. Faraday Soc., 31, 604 (1935). manuscript. 17) E. A. Moelwyn-Hughes and D. Glen, Roc. R. SOC.London, Ser. A,, 212, 260 (1952). References and Notes (18) C. D. Ritchie, “Solute-Solvent Interations”, J. F. Coetzee and C. 0. Ritchie, Eds., Marcel Dekker, New York, N.Y., 1969,Chapter 4,pp 266-268. (1) National Institutes of Health Fellow, 1967-1970. (19) R. A. Marcus, J. Phys. Chem., 72, 891 (1968). (2) R. P. Bell and D. M. Goodall, Proc. R. Soc.London, Ser. A, 294, 273 (1966); (20)Hydroxide (or alkoxide) ions are believed to be solvated by three tightly D. J. Barnes and R. P. Bell, ibid., 318,421 (1970);R. P. Bell and B. J. Cox, bound (inner sphere) water molecules. [The additional (outer sphere) water J. Chem. SOC.6, 194 (1970). molecules that are also present are not shown.] Judging from the energy (3) V. -

The Air Oxidation of 4,6-Di (2-Phenyl-2-Propyl) Pyrogallol. Spectroscopic and Kinetic Studies of the Intermediates
Carlsberg Res. Commun. Vol. 42, p. 11-25 THE AIR OXIDATION OF 4,6-DI (2-PHENYL-2-PROPYL) PYROGALLOL. SPECTROSCOPIC AND KINETIC STUDIES OF THE INTERMEDIATES. by HENRY I. ABRASH Department of Chemistry, Carlsberg Laboratory Gamle Carlsbergvej 10 - DK-2500 Copenhagen, Valby~ 1) Current address: Department of Chemistry, California State University at Northridge, Northridge, California 91324, USA Key words: 4,6-di (2-phenyl-2-propyl) pyrogallol, consecutive reactions, oxidation, oxygen, pH, quinone, semiquinone, spectra Two colored extinction bands, one with ~'max near 520 nm and the other near 770 nm, form and decay during air oxidations of 4,6-di(2-phenyl-2-propyl)pyrogallol in alkaline methanol. The 770 nm band appears to be due to the semiquinone monoanion while the extinction near 500 nm is thought to be due to both the semiquinone and 3-hydroxy-o-quinone. Although the rates of formation and decay of the two bands are indistinguishable in the presence of excess dissolved oxygen, the 770 nm band disappears slightly before the 500 nm extinction when the pyrogallol reactant is initially in excess of the dissolved oxygen. Except at high methoxide concentrations, the kinetics indicate a system of two consecutive reactions with pseudo first-order rate constants k~ and k 2. The pH' dependence of k~ indicates that the neutral pyrogallol, its monoanion and dianion all react, the rate increasing with the negative charge of the species. The k2/k~ ratio is constant at 5 from pH' 11 to 16. Extinction coefficients at both wavelengths increase with pH'. The 770 nm extinction disappears immediately after acidification while the 500 nm extinction decays at a rate consistent with oxidation. -

Ettore Majorana and the Birth of Autoionization
Ettore Majorana and the birth of autoionization E. Arimondo∗,† Charles W. Clark,‡ and W. C. Martin§ National Institute of Standards and Technology Gaithersburg, MD 20899, USA (Dated: May 19, 2009) Abstract In some of the first applications of modern quantum mechanics to the spectroscopy of many-electron atoms, Ettore Majorana solved several outstanding problems by developing the theory of autoionization. Later literature makes only sporadic refer- ences to this accomplishment. After reviewing his work in its contemporary context, we describe subsequent developments in understanding the spectra treated by Ma- jorana, and extensions of his theory to other areas of physics. We find many puzzles concerning the way in which the modern theory of autoionization was developed. ∗ Permanent address: Dipartimento di Fisica E. Fermi, Universit`adi Pisa, Italy †Electronic address: [email protected] ‡Electronic address: [email protected] §Electronic address: [email protected] 1 Contents I.Introduction 2 II.TheState of Atomic Spectroscopy circa 1931 4 A.Observed Spectra 4 B.Theoriesof Unstable Electronic States 6 III.Symmetry Considerations for Doubly-Excited States 7 IV.Analyses of the Observed Double-Excitation Spectra 8 A.Double Excitation in Helium 8 B.TheIncomplete np2 3P Terms in Zinc, Cadmium, and Mercury 9 V.Contemporary and Subsequent Work on Autoionization 12 A.Shenstone’s Contemporary Identification of Autoionization 12 B.Subsequent foundational work on autoionization 13 VI.Continuing Story of P − P0 Spectroscopy for Zinc, Cadmium, and Mercury 14 VII.Continuing Story of Double Excitation in Helium 16 VIII.Autoionization as a pervasive effect in physics 18 Acknowledgments 19 References 19 Figures 22 I. -

Solvent Deuterium Isotope Effect on Hydrolysis of Nd3+ Ion , and As Was
Notizen 1493 Solvent Deuterium Isotope Effect on Hydrolysis Experimental of Nd3+ Ion Reagents and apparatus A neodymium perchlorate sample solution and Mastjnobu Ma e d a *, T oshihiko A m ay a , and other reagents used were prepared and analyzed H id e t a k e K ak iha na according to the same procedures as those in the *Department of Applid Chemistry, previous papers5-6. Nagoya Institute of Technology, All the apparatus employed were the same as Gokiso, Showa-ku, Nagoya 466, Japan those in Ref. 7. Research Laboratory for Nuclear Reactors, Tokyo Institute of Technology, Preparation of a test solution O-okayama, Meguro-ku, Tokyo 152, Japan A test solution was prepared as follows. A (Z. Naturforsch. 32 b, 1493-1495 [1977]; received August 30, 1977) slightly acidic neodymium perchlorate solution, which had been freed from CO 2 by passing purified Deuterium Isotope Effect, Formation Constant, Heavy Water, Hydrolysis, Neodymium Ion N2 gas, was electrolyzed to reduce L+ ions by using a d. c. power supply until precipitates appeared. In order to saturate the solution with Nd(OL )3 precipi The hydrolysis equilibria of Nd3+ ion in tates the mixture was stirred with a magnetic rod light- and heavy-water solutions containing for two days. The precipitates of Nd(OL )3 were 3 mol dm -3 (Li)C104 as an ionic medium were removed by filtration with G 4 glass filters. All the studied at 25 °C by measuring the lyonium- procedures were carried out under an atmosphere ion concentration with a glass electrode. of N 2 gas in a room kept at 25 ± 1 °C. -

Auger Cascades in Resonantly Excited Neon
Auger cascades in resonantly excited neon masterarbeit zur Erlangung des akademischen Grades „Master of Science“ eingereicht bei der Physikalisch-Astronomischen Fakultät der Friedrich-Schiller-Universität Jena von Sebastian Stock geboren am 21. März 1993 in Aalen Jena, März 2017 Die in dieser Arbeit präsentierten Ergebnisse wurden ebenfalls in dem folgenden Artikel veröentlicht: S. Stock, R. Beerwerth, and S. Fritzsche. “Auger cascades in resonantly excited neon”. To be submitted. Erstgutachter: Prof. Dr. Stephan Fritzsche Zweitgutachter: Priv.-Doz. Dr. Andrey Volotka Abstract ¿e Auger cascades following the resonant 1s → 3p and 1s → 4p excitation of neutral neon are studied theoretically. In order to accurately predict Auger electron spectra, shake probabilities, ion yields, and the population of nal states, the complete cascade of decays from neutral to doubly-ionized neon is simulated by means of extensive mcdf calculations. Experimentally known values for the energy levels of neutral, singly and doubly ionized neon are utilized in order to further improve the simulated spectra. ¿e obtained results are compared to experimental ndings. For the most part, quite good agreement between theory and experiment is found. However, for the lifetime widths of certain energy levels of Ne+, larger dierences between the calculated values and the experiment are found. It is presumed that these discrepancies originate from the approximations that are utilized in the calculations of the Auger amplitudes. Contents 1 Introduction1 2 Overview of the Auger cascades3 3 Theory 7 3.1 Calculation of Auger amplitudes........................7 3.2 ¿e mcdf method.................................8 3.3 Shake processes and the biorthonormal transformation...........9 4 Calculations 11 4.1 Bound state wave function generation.................... -

Total of 10 Pages Only May Be Xeroxed
THE ACID CATALYZ D HYDROLYSIS OF M THYL ISOCYANIDE CENTRE FOR NEWFOUNDLAND STUDIES TOTAL OF 10 PAGES ONLY MAY BE XEROXED (Without Author's Permission) Y J Y lal V ( IM) 334618 THE ACID CATALYZED HYDROLYSIS OF METHYL ISOCYANIDE by © Yu-YUan Liew (Lim) A Thesis S'ubmi t ted to Memorial University of Newfoundland in partial fulfillment of the requirements for the degree of Master of Science Department of Chemistry May, 1972 ·.: '· ·. •, - i - ··.\., ACKNOWLEDGEMENT I wish to express my sju.::ere gratitude to Dr. A. R. Stein for his help, guidance and encouragement in making this investi- gation possible. I l-TOuld also like to thank Mr. D.H. Seymour for ::- the glass work, Drs. G. Ling and I. Mazurkiewicz (Department of Pharmacology, ~niversity of Ottawa) for their encouragement and help and my friends who helped make this thesis possible. I am also grateful to the National Research Council of Canada and Memorial University of Newfoundland for financial assistance. ii ·TAaLE .Of CONTENTS ACKNOWLEDGEMENT. • • • • • . • • • • • . • . • . • • . • . • . • • . • . • . • • • . • . • • • . i TABLE OF CONTENTS. • • • • • . • • . • . • • • . • . • • . • . • • . • • • . • . • • • . • • . • • . • • • . • • • • • . • i i LIST OF TABLES. • . i v LIST OF FIG'URES. • • • . • . • • • . • . • • . • . • . • . • • . • • . • . • . • . • vi ABSTRACT . .... .. ...................................... .. r • ~ ............... viii INTRODU~ .ION . ......•.•.•............ , • . • . • . 1 EXPERimNTAL. • . • . • . • . 13 1. General Considerations. • • • • • • • • • • -

SI Analytics Ph Handbook
pH handbook HELPFUL GUIDE FOR PRACTICAL APPLICATION AND USE Welcome to SI Analytics! We express our core competence, namely the production of analytical instruments, with our company name SI Analytics. SI also stands for the main products of our company: sensors and instruments. As part of the history of SCHOTT® AG, SI Analytics has more than 75 years experience in glass technology and in the development of analytical equipment. As always, our products are manufactured in Mainz with a high level of innovation and quality. Our electrodes, titrators and capillary viscometers will continue to be the right tools in any location where expertise in analytical measurement technology is required. In 2011 SI Analytics became part of the listed company Xylem Inc., headquartered in Rye Brook / N.Y., USA. Xylem is a leading international provider of water solutions. We look forward to presenting the pH handbook to you! The previous publication "Interesting facts about pH measurement" has been restructured, made clearer and more engaging, and extra information has been added. The focus has been consciously put on linking general information with our lab findings and making this accessible to you in a practical format. Complemented by the reference to our range of products and with practical recommendations for use with specific applications, the pH primer is an indispensable accompaniment for everyday lab work. We at SI Analytics would be happy to work successfully together with you in the future. SI Analytics GmbH Dr. Robert Reining CEO CONTENTS SECTION 1 Basic concepts of potentiometry and pH measurement 1.1 Definition of acid and base ......................................................... -

Xerox University Microfilms 300 North Zeeb Rood Ann Arbor, Michigan 48106 11
INFORMATION TO USERS This material was produced from a microfilm copy of the original document. While the most advanced technological means to photograph and reproduce this document have been used, the quality is heavily dependent upon the quality of the original submitted. The following explanation of techniques is provided to help you understand markings or patterns which may appear on this reproduction. 1. The sign or "target" for pages apparently lacking from the document photographed is "Missing Page(s)". If it was possible to obtain the missing page(s) or section, they are spliced into the film along with adjacent pages. This may have necessitated cutting thru an image and duplicating adjacent pages to insure you complete continuity. 2. When an image^pn the film is obliterated with a large round black mark, it is an indication that the photographer suspected that the copy may have moved during exposure and thus cause a blurred image. You will find a good image of the page in the adjacent frame. 3. When a map, drawing or chart, etc., was part of the material being photographed the photographer followed a definite method in "sectioning" the material. It is customary to begin photoing at the upper left hand corner of a large sheet and to continue photoing from left to right in equal sections with a small overlap. If necessary, sectioning is continued again — beginning below the first row and continuing on until complete. 4. The majority of users indicate that the textual content is of greatest value, however, a somewhat higher quality reproduction could be made from "photographs" if essential to the understanding of the dissertation. -

Redox-Driven Conformational Dynamics in a Photosystem-II-Inspired
Redox-driven Conformational Dynamics in a Photosystem-II-inspired β-hairpin Maquette Determined through Spectroscopy and Simulation Hyea Hwang†,1, Tyler G. McCaslin†,2,3 Anthony Hazel4, Cynthia V. Pagba2,3 Christina M. ∗ Nevin2,3, Anna Pavlova4, Bridgette A. Barry2,3 and James C. Gumbart2,3,4, 1School of Materials Science and Engineering, 2School of Chemistry and Biochemistry, 3Petit Institute for Bioengineering and Biosciences, and 4School of Physics, Georgia Institute of Technology, Atlanta, GA 30332 † These authors contributed equally to this work. ∗ To whom correspondence should be addressed; Email: [email protected]; Phone: 404-385-0797 1 Abstract Tyrosine-based radical transfer plays an important role in photosynthesis, respiration, and DNA synthesis. Radical transfer can occur either by electron transfer (ET) or proton coupled electron transfer (PCET), depending on the pH. Reversible conformational changes in the surrounding protein matrix may control reactivity of radical intermediates. De novo designed Peptide A is a synthetic 18 amino-acid β-hairpin, which contains a single tyrosine (Y5) and carries out a kinetically significant PCET reaction between Y5 and a cross-strand histidine (H14). In Peptide A, amide II’ (CN) changes are observed in the UV resonance Raman (UVRR) spectrum, associated with tyrosine ET and PCET; these bands were attributed previously to a reversible change in secondary structure. Here, we use molecular dynamics simulations to define this conformational change in Peptide A and its H14-to-cyclohexylalanine variant, Peptide C. Three different Y5 charge states, tyrosine (YH), tyrosinate (Y–), and neutral tyrosyl radical (Y•), are considered. The simulations show that Peptide A-YH and A-Y– retain secondary structure and noncovalent interactions, whereas A-Y• is unstable. -

Angular Distribution of Autoionization and Auger Electrons Ejected by Electron Impact from Laser-Excited and Polarized Atoms
J. Phys. B: At. Mol. Opt. Phys. 30 (1997) 1269–1291. Printed in the UK PII: S0953-4075(97)76979-7 Angular distribution of autoionization and Auger electrons ejected by electron impact from laser-excited and polarized atoms V V Balashov, A N Grum-Grzhimailo and N M Kabachnik Institute of Nuclear Physics, Moscow State University, Moscow 119899, Russia Received 8 August 1996, in final form 8 November 1996 Abstract. A general formalism is developed for a description of the angular distribution of electrons ejected after the electron-impact excitation/ionization of the laser-excited and polarized atoms. Properties of the angular distributions are analysed in a general case as well as in some particular geometries of the experiment. Within the two-step approximation the formalism is applied to the resonant single and double ionization with ejection of autoionization and Auger electrons. As an example the angular distributions of autoionization electrons from laser- excited sodium atoms are calculated in the plane-wave and distorted-wave Born approximations. Angular correlations between autoionization and scattered electrons, which can be measured in a coincidence (e, 2e) experiment with polarized targets, are also analysed. 1. Introduction The study of angular distributions of electrons ejected from atoms in electron–atom collisions is a well developed field which gives important information on mechanisms of excitation and decay of atomic quasistationary states (Mehlhorn 1985, 1990). In the majority of the studies, both experimental and theoretical, the electron-impact excitation from the ground states of atoms was investigated. Ten years ago Balashov and Grum-Grzhimailo (1986) suggested the investigation of the electron-impact excitation of atomic autoionizing states in atoms excited by lasers. -

Ultrafast Resonant Interatomic Coulombic Decay Induced by Quantum Fluid Dynamics
PHYSICAL REVIEW X 11, 021011 (2021) Ultrafast Resonant Interatomic Coulombic Decay Induced by Quantum Fluid Dynamics A. C. LaForge ,1,2,* R. Michiels,2 Y. Ovcharenko ,3,§ A. Ngai,2 J. M. Escartín ,4 N. Berrah ,1 C. Callegari ,5 A. Clark,6 M. Coreno,7 R. Cucini,5 M. Di Fraia,5 M. Drabbels,6 E. Fasshauer,8 P. Finetti,5 L. Giannessi,5,9 C. Grazioli ,18 D. Iablonskyi,10 B. Langbehn ,3 T. Nishiyama,11 V. Oliver,6 P. Piseri,12 O. Plekan,5 K. C. Prince ,5 D. Rupp,3,¶ S. Stranges ,13 K. Ueda,10 N. Sisourat,14 J. Eloranta,15 M. Pi,16,17 M. Barranco,16,17 F. Stienkemeier ,2 † ‡ T. Möller,3, and M. Mudrich8, 1Department of Physics, University of Connecticut, Storrs, Connecticut 06269, USA 2Institute of Physics, University of Freiburg, 79104 Freiburg, Germany 3Institut für Optik und Atomare Physik, Technische Universität Berlin, 10623 Berlin, Germany 4Institut de Química Teòrica i Computacional, Universitat de Barcelona, Carrer de Martí i Franqu`es 1, 08028 Barcelona, Spain 5Elettra-Sincrotrone Trieste, 34149 Basovizza, Trieste, Italy 6Laboratoire Chimie Physique Mol´eculaire, Ecole Polytechnique F´ed´erale de Lausanne, 1015 Lausanne, Switzerland 7CNR, Istituto di Struttura della Materia, 00016 Monterotondo Scalo, Italy 8Department of Physics and Astronomy, Aarhus University, 8000 Aarhus C, Denmark 9Nazionale di Fisica Nucleare, Laboratori Nazionali di Frascati, Via E. Fermi 40, 00044 Frascati, Roma, Italy CNR-Istituto Officina dei Materiali, Laboratorio TASC, 34149 Trieste, Italy 10Institute of Multidisciplinary Research for Advanced Materials, -
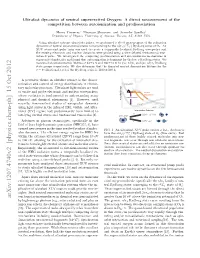
Ultrafast Dynamics of Neutral Superexcited Oxygen: a Direct Measurement of the Competition Between Autoionization and Predissociation
Ultrafast dynamics of neutral superexcited Oxygen: A direct measurement of the competition between autoionization and predissociation Henry Timmers,∗ Niranjan Shivaram, and Arvinder Sandhuy Department of Physics, University of Arizona, Tucson, AZ, 85721 USA. Using ultrafast extreme ultraviolet pulses, we performed a direct measurement of the relaxation 4 − dynamics of neutral superexcited states corresponding to the nlσg(c Σu ) Rydberg series of O2. An XUV attosecond pulse train was used to create a temporally localized Rydberg wavepacket and the ensuing electronic and nuclear dynamics were probed using a time-delayed femtosecond near- infrared pulse. We investigated the competing predissociation and autoionization mechanisms in superexcited molecules and found that autoionization is dominant for the low n Rydberg states. We measured an autoionization lifetime of 92±6 fs and 180±10 fs for (5s; 4d)σg and (6s; 5d)σg Rydberg state groups respectively. We also determine that the disputed neutral dissociation lifetime for the ν = 0 vibrational level of the Rydberg series is 1100 ± 100 fs. A pervasive theme in ultrafast science is the charac- terization and control of energy distributions in elemen- tary molecular processes. Ultrashort light pulses are used Energy to excite and probe electronic and nuclear wavepackets, Neutral whose evolution is fundamental to understanding many Dissociation Autoionization physical and chemical phenomena [1]. However, until Potential recently, time-resolved studies of wavepacket dynamics using light pulses in the infrared (IR), visible, and ultra- violet (UV) regime had predominantly been limited to Internuclear Distance low-lying excited states and femtosecond timescales [2]. Advances in photon technologies, specifically in the ( )+ field of laser high-harmonic generation (HHG)[3, 4], have opened new avenues in the time-resolved studies of molec- FIG.