Increasing Accuracy of Powdery Mildew (Ascomycota, Erysiphales) Identification Using Previously Untapped DNA Regions
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

New Powdery Mildew on Tomatoes
NEW POWDERY MILDEW ON TOMATOES Heather Scheck, Plant Pathologist Ag Commissioner’s Office, Santa Barbara County POWDERY MILDEW BIOLOGY Powdery mildew fungi are obligate, biotrophic parasites of the phylum Ascomycota of the Kingdom Fungi. The diseases they cause are common, widespread, and easily recognizable Individual species of powdery mildew fungi typically have a narrow host range, but the ones that infect Tomato are exceptionally large. Photo from APS Net POWDERY MILDEW BIOLOGY Unlike most fungal pathogens, powdery mildew fungi tend to grow superficially, or epiphytically, on plant surfaces. During the growing season, hyphae and spores are produced in large colonies that can coalesce Infections can also occur on stems, flowers, or fruit (but not tomato fruit) Our climate allows easy overwintering of inoculum and perfect summer temperatures for epidemics POWDERY MILDEW BIOLOGY Specialized absorption cells, termed haustoria, extend into the plant epidermal cells to obtain nutrition. Powdery mildew fungi can completely cover the exterior of the plant surfaces (leaves, stems, fruit) POWDERY MILDEW BIOLOGY Conidia (asexual spores) are also produced on plant surfaces during the growing season. The conidia develop either singly or in chains on specialized hyphae called conidiophores. Conidiophores arise from the epiphytic hyphae. This is the Anamorph. Courtesy J. Schlesselman POWDERY MILDEW BIOLOGY Some powdery mildew fungi produce sexual spores, known as ascospores, in a sac-like ascus, enclosed in a fruiting body called a chasmothecium (old name cleistothecium). This is the Teleomorph Chasmothecia are generally spherical with no natural opening; asci with ascospores are released when a crack develops in the wall of the fruiting body. -

Additions to the Turkish Discomycetes
Turkish Journal of Botany Turk J Bot (2014) 38: 617-622 http://journals.tubitak.gov.tr/botany/ © TÜBİTAK Research Note doi:10.3906/bot-1307-53 Additions to the Turkish Discomycetes 1, 2 2 3 Yusuf UZUN *, İsmail ACAR , Mustafa Emre AKÇAY , Ilgaz AKATA 1 Department of Pharmaceutical Sciences, Faculty of Pharmacy, Yüzüncü Yıl University, Van, TURKEY 2 Department of Biology, Faculty of Science, Yüzüncü Yıl University, Van, Turkey 3 Department of Biology, Faculty of Science, Ankara University, Ankara, Turkey Received: 20.07.2013 Accepted: 23.01.2014 Published Online: 31.03.2014 Printed: 30.04.2014 Abstract: In this study, Mollisia ventosa P.Karst., Hymenoscyphus herbarum (Pers.) Dennis, H. immutabilis (Fuckel) Dennis, H. robustior (P.Karst.) Dennis, Ciboria amentacea (Balb.) Fuckel, and Peziza fimeti (Fuckel) E.C.Hansen have been recorded in Turkey for the first time. Comments on their morphological and anatomical features, geographical position, locality, collection date, and distribution and short descriptions and photographs related to macro- and micromorphologies of the taxa are provided. Key words: Biodiversity, Discomycetes, new records, Turkey 1. Introduction detailed studies. Distilled water, Melzer’s reagent, and 5% The apothecial ascomycetes that produce cup-shaped KOH were used for microscopic investigation. ascomata with various colors are commonly known as Microphotographs of apothecia were taken under a discomycetes. They represent approximately 9000 taxa, light microscope (Leica DM 1000). The specimens were which are saprobic, parasitic, mycorrhizal, or lichenized. identified with the help of Breitenbach and Kränzlin Some discomycetes are used in drugs or as biological (1984), Hansen and Knudsen (2000), Spooner (2001), control agents. -

Psychrophilic Fungi from the World's Roof
Persoonia 34, 2015: 100–112 www.ingentaconnect.com/content/nhn/pimj RESEARCH ARTICLE http://dx.doi.org/10.3767/003158515X685878 Psychrophilic fungi from the world’s roof M. Wang1,2, X. Jiang3, W. Wu3, Y. Hao1, Y. Su1, L. Cai1, M. Xiang1, X. Liu1 Key words Abstract During a survey of cold-adapted fungi in alpine glaciers on the Qinghai-Tibet Plateau, 1 428 fungal isolates were obtained of which 150 species were preliminary identified. Phoma sclerotioides and Pseudogymnoascus pan- glaciers norum were the most dominant species. Psychrotolerant species in Helotiales (Leotiomycetes, Ascomycota) were Phoma sclerotioides studied in more detail as they represented the most commonly encountered group during this investigation. Two Pseudogymnoascus pannorum phylogenetic trees were constructed based on the partial large subunit nrDNA (LSU) to infer the taxonomic place- Psychrophila ments of these strains. Our strains nested in two well-supported major clades, which represented Tetracladium and psychrotolerant a previously unknown lineage. The unknown lineage is distant to any other currently known genera in Helotiales. Tetracladium Psychrophila gen. nov. was therefore established to accommodate these strains which are characterised by globose or subglobose conidia formed from phialides on short or reduced conidiophores. Our analysis also showed that an LSU-based phylogeny is insufficient in differentiating strains at species level. Additional analyses using combined sequences of ITS+TEF1+TUB regions were employed to further investigate the phylogenetic relationships of these strains. Together with the recognisable morphological distinctions, six new species (i.e. P. antarctica, P. lutea, P. oli- vacea, T. ellipsoideum, T. globosum and T. psychrophilum) were described. Our preliminary investigation indicates a high diversity of cold-adapted species in nature, and many of them may represent unknown species. -

The Culture-Independent Analysis of Fungal Endophytes of Wheat Grown in Kwazulu-Natal, South Africa
THE CULTURE-INDEPENDENT ANALYSIS OF FUNGAL ENDOPHYTES OF WHEAT GROWN IN KWAZULU-NATAL, SOUTH AFRICA By Richard Jörn Burgdorf Submitted in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY In Microbiology School of Life Sciences College of Agriculture, Engineering and Science Pietermaritzburg South Africa December 2016 Thesis summary Fungal endophytes are of interest due to their diverse taxonomy and biological functions. A range of definitions exists based on their identity, morphology, location and relationship with their host. Fungal endophytes belong to a wide range of taxa and they are categorized by a variety of characteristics. The detection and identification of these fungal endophytes can be performed using culture-dependent and culture-independent methods. These organisms have a range of application in pharmaceutical discovery and agriculture. Agricultural applications include the exploitation of the growth promoting and protective properties of fungal endophytes in crops such as wheat. This important crop is grown in South Africa where biotic and environmental stresses pose a challenge to its cultivation. Fungal endophytes have demonstrated potential to ameliorate these challenges. Future research will reveal how they can be harnessed to fight food insecurity brought about by stress factors such as climate change. Extraneous DNA interferes with PCR studies of endophytic fungi. A procedure was developed with which to evaluate the removal of extraneous DNA. Wheat (Triticum aestivum) leaves were sprayed with Saccharomyces cerevisiae and then subjected to physical and chemical surface treatments. The fungal ITS1 products were amplified from whole tissue DNA extractions. ANOVA was performed on the DNA bands representing S. cerevisiae on the agarose gel. -

Basal Resistance of Barley to Adapted and Non-Adapted Forms of Blumeria Graminis
Basal resistance of barley to adapted and non-adapted forms of Blumeria graminis Reza Aghnoum Thesis committee Thesis supervisors Prof. Dr. Richard G.F. Visser Professor of Plant Breeding Wageningen University Dr.ir. Rients E. Niks Assistant professor, Laboratory of Plant Breeding Wageningen University Other members Prof. Dr. R.F. Hoekstra, Wageningen University Prof. Dr. F. Govers, Wageningen University Prof. Dr. ir. C. Pieterse, Utrecht University Dr.ir. G.H.J. Kema, Plant Research International, Wageningen This research was conducted under the auspices of the Graduate school of Experimental Plant Sciences. II Basal resistance of barley to adapted and non-adapted forms of Blumeria graminis Reza Aghnoum Thesis Submitted in partial fulfillment of the requirements for the degree of doctor at Wageningen University by the authority of the Rector Magnificus Prof. Dr. M.J. Kropff, in the presence of the Thesis Committee appointed by the Doctorate Board to be defended in public on Tuesday 16 June 2009 at 4 PM in the Aula. III Reza Aghnoum Basal resistance of barley to adapted and non-adapted forms of Blumeria graminis 132 pages. Thesis, Wageningen University, Wageningen, NL (2009) With references, with summaries in Dutch and English ISBN 978-90-8585-419-7 IV Contents Chapter 1 1 General introduction Chapter 2 15 Which candidate genes are responsible for natural variation in basal resistance of barley to barley powdery mildew? Chapter 3 47 Transgressive segregation for extreme low and high level of basal resistance to powdery mildew in barley -

First Records of Two Ascomycete Fungi (Ascomycota) for Slovenia
NATURA SLOVENIAE 21(2): 5-11 Prejeto / Received: 5. 6. 2019 SHORT COMMUNICATION Sprejeto / Accepted: 30. 12. 2019 First records of two ascomycete fungi (Ascomycota) for Slovenia Luka ŠPARL1, Eva ZUPAN2 1Služba Krajinski park Tivoli, Rožnik in Šišenski hrib, Javno podjetje VOKA Snaga d. o. o., Vodovodna cesta 90, SI-1001 Ljubljana; E-mail: [email protected] 2Eva ZUPAN, Vrhovci c XII/24a, SI-1000 Ljubljana; E-mail: [email protected] Abstract. In April and May 2019, two ascomycetous species – Vibrissea filisporia (Bonord.) Korf & A. Sánchez 1967 and Cudoniella tenuispora (Cooke & Massee) Dennis 1974 were observed in Tivoli, Rožnik and Šiška hill Landscape Park in central Slovenia. This is the first evidence of their presence in the country. Despite specific growth condition requirements, there is a reasonable probability that these two species grow also elsewhere in Slovenia, but have simply been overlooked. We recommend further studies of suitable habitats for the species, to complete the knowledge on their distribution within the country. Key words: fungi, Ascomycota, first records, Vibrissea filisporia, Cudoniella tenuispora, Mali Rožnik, Ljubljana, Slovenia Izvleček. Prvi podatki o dveh vrstah gliv zaprtotrosnic (Ascomycota) za Slovenijo – V aprilu in maju 2019 sta bili na območju zavarovanega območja Krajinskega parka Tivoli, Rožnik in Šišenski hrib v osrednji Sloveniji najdeni dve glivi zaprtotrosnici, Vibrissea filisporia (Bonord.) Korf & A. Sánchez 1967 in Cudoniella tenuispora (Cooke & Massee) Dennis 1974. Kljub specifičnim rastiščnim zahtevam je zelo verjetno, da ti dve vrsti uspevata tudi drugod po državi, a sta bili spregledani. Prihodnje študije primernih habitatov bodo lahko dopolnile poznavanje razširjenosti teh vrst v državi. -

Characterization of the Ergosterol Biosynthesis Pathway in Ceratocystidaceae
Journal of Fungi Article Characterization of the Ergosterol Biosynthesis Pathway in Ceratocystidaceae Mohammad Sayari 1,2,*, Magrieta A. van der Nest 1,3, Emma T. Steenkamp 1, Saleh Rahimlou 4 , Almuth Hammerbacher 1 and Brenda D. Wingfield 1 1 Department of Biochemistry, Genetics and Microbiology, Forestry and Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria 0002, South Africa; [email protected] (M.A.v.d.N.); [email protected] (E.T.S.); [email protected] (A.H.); brenda.wingfi[email protected] (B.D.W.) 2 Department of Plant Science, University of Manitoba, 222 Agriculture Building, Winnipeg, MB R3T 2N2, Canada 3 Biotechnology Platform, Agricultural Research Council (ARC), Onderstepoort Campus, Pretoria 0110, South Africa 4 Department of Mycology and Microbiology, University of Tartu, 14A Ravila, 50411 Tartu, Estonia; [email protected] * Correspondence: [email protected]; Fax: +1-204-474-7528 Abstract: Terpenes represent the biggest group of natural compounds on earth. This large class of organic hydrocarbons is distributed among all cellular organisms, including fungi. The different classes of terpenes produced by fungi are mono, sesqui, di- and triterpenes, although triterpene ergosterol is the main sterol identified in cell membranes of these organisms. The availability of genomic data from members in the Ceratocystidaceae enabled the detection and characterization of the genes encoding the enzymes in the mevalonate and ergosterol biosynthetic pathways. Using Citation: Sayari, M.; van der Nest, a bioinformatics approach, fungal orthologs of sterol biosynthesis genes in nine different species M.A.; Steenkamp, E.T.; Rahimlou, S.; of the Ceratocystidaceae were identified. -
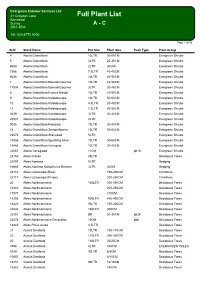
Full Plant List Banstead Surrey a - C SM7 3BW
Evergreen Exterior Services Ltd 21 Croydon Lane Full Plant List Banstead Surrey A - C SM7 3BW Tel: 020 8770 9200 Page 1 of 56 SLN Stock NamePot Size Plant Size Pack Type Plant Group 4 Abelia Grandiflora 10LTR 50-60CM Evergreen Shrubs 5 Abelia Grandiflora 3LTR 25-30CM Evergreen Shrubs 5680 Abelia Grandiflora 2LTR 30CM+ Evergreen Shrubs 7366 Abelia Grandiflora 7.5LTR 40-45CM Evergreen Shrubs 9036 Abelia Grandiflora 10LTR 40-50CM Evergreen Shrubs 1 Abelia Grandiflora Edward Goucher 10LTR 40-50CM Evergreen Shrubs 17934 Abelia Grandiflora Edward Goucher 3LTR 30-40CM Evergreen Shrubs 8 Abelia Grandiflora Francis Mason 10LTR 40-50CM Evergreen Shrubs 9 Abelia Grandiflora Kaleidoscope 10LTR 50-60CM Evergreen Shrubs 10 Abelia Grandiflora Kaleidoscope 4.5LTR 30-40CM Evergreen Shrubs 11 Abelia Grandiflora Kaleidoscope 7.5LTR 40-50CM Evergreen Shrubs 3639 Abelia Grandiflora Kaleidoscope 3LTR 30-40CM Evergreen Shrubs 23531 Abelia Grandiflora Kaleidoscope 2LTR Evergreen Shrubs 9035 Abelia Grandiflora Prostrata 10LTR 30-40CM Evergreen Shrubs 14 Abelia Grandiflora Semperflorens 10LTR 50-60CM Evergreen Shrubs 22072 Abelia Grandiflora Sherwood 5LTR Evergreen Shrubs 15956 Abelia Grandiflora Sparkling Silver 10LTR 50-60CM Evergreen Shrubs 15486 Abelia Grandiflora Variegata 10LTR 30-40CM Evergreen Shrubs 22657 Abelia Variegated 11CM pk10 Evergreen Shrubs 23153 Abies Fraseri 45LTR Deciduous Trees 22097 Abies Koreana 5LTR Hedging 16665 Abies Koreana Kohout's Ice Breaker 3LTR 00CM Hedging 22116 Abies Lasiocarpa (Blue) 150-200CM Christmas 22117 Abies Lasiocarpa -

Recently Introduced Diseases of Ornamental Plants Gardens Are Being Increasingly Exposed to New Diseases, Principally Due to Globalization of Plant Trade
horticultural science Recently introduced diseases of ornamental plants Gardens are being increasingly exposed to new diseases, principally due to globalization of plant trade. Béatrice Henricot surveys the most significant introductions he main reasons for One key question is whether new and infamous examples include increased risks of introducing we are recording more new diseases potato blight in the 19th century and Tpathogens into any country than in the past as a result of these Dutch elm disease in the late 1960s. are globalization of trade and travel, factors. A study carried out by Jones More recently, gardeners have had evolutionary change of plant patho & Baker (2007) indicated that the to deal with box blight and new gens, and climate change and in part numbers of new or important species of Phytophthora such as icular its effect on insects as disease pathogens estab lishing in the UK P. ramorum and P. kernoviae. vectors (Waage et al. 2007). Global during the period 1970–2004 are This article will look at diseases ization is probably the main driver of not increasing. A total of 234 new which have recently been found increased risk of intro ducing plant pathogens were recorded during that in gardens in the UK. Cases mainly pathogens in the future and this has period and were mainly associated come from the RHS advisory been well documented (Brasier 2005, with ornamental plants (as opposed records since 1998 with some Henricot & Gorton 2005, Ingram to horticultural crops, wild native additions taken from the online 2005, Brasier 2008). Pathogen evol species, agricult ural crops, pasture journal New Disease Reports (www. -

Morinagadepsin, a Depsipeptide from the Fungus Morinagamyces Vermicularis Gen. Et Comb. Nov
microorganisms Article Morinagadepsin, a Depsipeptide from the Fungus Morinagamyces vermicularis gen. et comb. nov. Karen Harms 1,2 , Frank Surup 1,2,* , Marc Stadler 1,2 , Alberto Miguel Stchigel 3 and Yasmina Marin-Felix 1,* 1 Department Microbial Drugs, Helmholtz Centre for Infection Research, Inhoffenstrasse 7, 38124 Braunschweig, Germany; [email protected] (K.H.); [email protected] (M.S.) 2 Institute of Microbiology, Technische Universität Braunschweig, Spielmannstrasse 7, 38106 Braunschweig, Germany 3 Mycology Unit, Medical School and IISPV, Universitat Rovira i Virgili, C/ Sant Llorenç 21, 43201 Reus, Tarragona, Spain; [email protected] * Correspondence: [email protected] (F.S.); [email protected] (Y.M.-F.) Abstract: The new genus Morinagamyces is introduced herein to accommodate the fungus Apiosordaria vermicularis as inferred from a phylogenetic study based on sequences of the internal transcribed spacer region (ITS), the nuclear rDNA large subunit (LSU), and partial fragments of ribosomal polymerase II subunit 2 (rpb2) and β-tubulin (tub2) genes. Morinagamyces vermicularis was analyzed for the production of secondary metabolites, resulting in the isolation of a new depsipeptide named morinagadepsin (1), and the already known chaetone B (3). While the planar structure of 1 was elucidated by extensive 1D- and 2D-NMR analysis and high-resolution mass spectrometry, the absolute configuration of the building blocks Ala, Val, and Leu was determined as -L by Marfey’s method. The configuration of the 3-hydroxy-2-methyldecanyl unit was assigned as 22R,23R by Citation: Harms, K.; Surup, F.; Stadler, M.; Stchigel, A.M.; J-based configuration analysis and Mosher’s method after partial hydrolysis of the morinagadepsin Marin-Felix, Y. -

Ceratocystidaceae Exhibit High Levels of Recombination at the Mating-Type (MAT) Locus
Accepted Manuscript Ceratocystidaceae exhibit high levels of recombination at the mating-type (MAT) locus Melissa C. Simpson, Martin P.A. Coetzee, Magriet A. van der Nest, Michael J. Wingfield, Brenda D. Wingfield PII: S1878-6146(18)30293-9 DOI: 10.1016/j.funbio.2018.09.003 Reference: FUNBIO 959 To appear in: Fungal Biology Received Date: 10 November 2017 Revised Date: 11 July 2018 Accepted Date: 12 September 2018 Please cite this article as: Simpson, M.C., Coetzee, M.P.A., van der Nest, M.A., Wingfield, M.J., Wingfield, B.D., Ceratocystidaceae exhibit high levels of recombination at the mating-type (MAT) locus, Fungal Biology (2018), doi: https://doi.org/10.1016/j.funbio.2018.09.003. This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain. ACCEPTED MANUSCRIPT 1 Title 2 Ceratocystidaceae exhibit high levels of recombination at the mating-type ( MAT ) locus 3 4 Authors 5 Melissa C. Simpson 6 [email protected] 7 Martin P.A. Coetzee 8 [email protected] 9 Magriet A. van der Nest 10 [email protected] 11 Michael J. Wingfield 12 [email protected] 13 Brenda D. -

Molecular Identification of Fungi
Molecular Identification of Fungi Youssuf Gherbawy l Kerstin Voigt Editors Molecular Identification of Fungi Editors Prof. Dr. Youssuf Gherbawy Dr. Kerstin Voigt South Valley University University of Jena Faculty of Science School of Biology and Pharmacy Department of Botany Institute of Microbiology 83523 Qena, Egypt Neugasse 25 [email protected] 07743 Jena, Germany [email protected] ISBN 978-3-642-05041-1 e-ISBN 978-3-642-05042-8 DOI 10.1007/978-3-642-05042-8 Springer Heidelberg Dordrecht London New York Library of Congress Control Number: 2009938949 # Springer-Verlag Berlin Heidelberg 2010 This work is subject to copyright. All rights are reserved, whether the whole or part of the material is concerned, specifically the rights of translation, reprinting, reuse of illustrations, recitation, broadcasting, reproduction on microfilm or in any other way, and storage in data banks. Duplication of this publication or parts thereof is permitted only under the provisions of the German Copyright Law of September 9, 1965, in its current version, and permission for use must always be obtained from Springer. Violations are liable to prosecution under the German Copyright Law. The use of general descriptive names, registered names, trademarks, etc. in this publication does not imply, even in the absence of a specific statement, that such names are exempt from the relevant protective laws and regulations and therefore free for general use. Cover design: WMXDesign GmbH, Heidelberg, Germany, kindly supported by ‘leopardy.com’ Printed on acid-free paper Springer is part of Springer Science+Business Media (www.springer.com) Dedicated to Prof. Lajos Ferenczy (1930–2004) microbiologist, mycologist and member of the Hungarian Academy of Sciences, one of the most outstanding Hungarian biologists of the twentieth century Preface Fungi comprise a vast variety of microorganisms and are numerically among the most abundant eukaryotes on Earth’s biosphere.