R Stuck Thesis Final Document May 2017
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Common Names for Selected Aromatic Groups
More Nomenclature: Common Names for Selected Aromatic Groups Phenyl group = or Ph = C6H5 = Aryl = Ar = aromatic group. It is a broad term, and includes any aromatic rings. Benzyl = Bn = It has a -CH2- (methylene) group attached to the benzene ring. This group can be used to name particular compounds, such as the one shown below. This compound has chlorine attached to a benzyl group, therefore it is called benzyl chloride. Benzoyl = Bz = . This is different from benzyl group (there is an extra “o” in the name). It has a carbonyl attached to the benzene ring instead of a methylene group. For example, is named benzoyl chloride. Therefore, it is sometimes helpful to recognize a common structure in order to name a compound. Example: Nomenclature: 3-phenylpentane Example: This is Amaize. It is used to enhance the yield of corn production. The systematic name for this compound is 2,4-dinitro-6-(1-methylpropyl)phenol. Polynuclear Aromatic Compounds Aromatic rings can fuse together to form polynuclear aromatic compounds. Example: It is two benzene rings fused together, and it is aromatic. The electrons are delocalized in both rings (think about all of its resonance form). Example: This compound is also aromatic, including the ring in the middle. All carbons are sp2 hybridized and the electron density is shared across all 5 rings. Example: DDT is an insecticide and helped to wipe out malaria in many parts of the world. Consequently, the person who discovered it (Muller) won the Nobel Prize in 1942. The systematic name for this compound is 1,1,1-trichloro-2,2-bis-(4-chlorophenyl)ethane. -

Organic Chemistry
Wisebridge Learning Systems Organic Chemistry Reaction Mechanisms Pocket-Book WLS www.wisebridgelearning.com © 2006 J S Wetzel LEARNING STRATEGIES CONTENTS ● The key to building intuition is to develop the habit ALKANES of asking how each particular mechanism reflects Thermal Cracking - Pyrolysis . 1 general principles. Look for the concepts behind Combustion . 1 the chemistry to make organic chemistry more co- Free Radical Halogenation. 2 herent and rewarding. ALKENES Electrophilic Addition of HX to Alkenes . 3 ● Acid Catalyzed Hydration of Alkenes . 4 Exothermic reactions tend to follow pathways Electrophilic Addition of Halogens to Alkenes . 5 where like charges can separate or where un- Halohydrin Formation . 6 like charges can come together. When reading Free Radical Addition of HX to Alkenes . 7 organic chemistry mechanisms, keep the elec- Catalytic Hydrogenation of Alkenes. 8 tronegativities of the elements and their valence Oxidation of Alkenes to Vicinal Diols. 9 electron configurations always in your mind. Try Oxidative Cleavage of Alkenes . 10 to nterpret electron movement in terms of energy Ozonolysis of Alkenes . 10 Allylic Halogenation . 11 to make the reactions easier to understand and Oxymercuration-Demercuration . 13 remember. Hydroboration of Alkenes . 14 ALKYNES ● For MCAT preparation, pay special attention to Electrophilic Addition of HX to Alkynes . 15 Hydration of Alkynes. 15 reactions where the product hinges on regio- Free Radical Addition of HX to Alkynes . 16 and stereo-selectivity and reactions involving Electrophilic Halogenation of Alkynes. 16 resonant intermediates, which are special favor- Hydroboration of Alkynes . 17 ites of the test-writers. Catalytic Hydrogenation of Alkynes. 17 Reduction of Alkynes with Alkali Metal/Ammonia . 18 Formation and Use of Acetylide Anion Nucleophiles . -

Aldehydes and Ketones
12 Aldehydes and Ketones Ethanol from alcoholic beverages is first metabolized to acetaldehyde before being broken down further in the body. The reactivity of the carbonyl group of acetaldehyde allows it to bind to proteins in the body, the products of which lead to tissue damage and organ disease. Inset: A model of acetaldehyde. (Novastock/ Stock Connection/Glow Images) KEY QUESTIONS 12.1 What Are Aldehydes and Ketones? 12.8 What Is Keto–Enol Tautomerism? 12.2 How Are Aldehydes and Ketones Named? 12.9 How Are Aldehydes and Ketones Oxidized? 12.3 What Are the Physical Properties of Aldehydes 12.10 How Are Aldehydes and Ketones Reduced? and Ketones? 12.4 What Is the Most Common Reaction Theme of HOW TO Aldehydes and Ketones? 12.1 How to Predict the Product of a Grignard Reaction 12.5 What Are Grignard Reagents, and How Do They 12.2 How to Determine the Reactants Used to React with Aldehydes and Ketones? Synthesize a Hemiacetal or Acetal 12.6 What Are Hemiacetals and Acetals? 12.7 How Do Aldehydes and Ketones React with CHEMICAL CONNECTIONS Ammonia and Amines? 12A A Green Synthesis of Adipic Acid IN THIS AND several of the following chapters, we study the physical and chemical properties of compounds containing the carbonyl group, C O. Because this group is the functional group of aldehydes, ketones, and carboxylic acids and their derivatives, it is one of the most important functional groups in organic chemistry and in the chemistry of biological systems. The chemical properties of the carbonyl group are straightforward, and an understanding of its characteristic reaction themes leads very quickly to an understanding of a wide variety of organic reactions. -
![Solvent Effects on Structure and Reaction Mechanism: a Theoretical Study of [2 + 2] Polar Cycloaddition Between Ketene and Imine](https://docslib.b-cdn.net/cover/9440/solvent-effects-on-structure-and-reaction-mechanism-a-theoretical-study-of-2-2-polar-cycloaddition-between-ketene-and-imine-159440.webp)
Solvent Effects on Structure and Reaction Mechanism: a Theoretical Study of [2 + 2] Polar Cycloaddition Between Ketene and Imine
J. Phys. Chem. B 1998, 102, 7877-7881 7877 Solvent Effects on Structure and Reaction Mechanism: A Theoretical Study of [2 + 2] Polar Cycloaddition between Ketene and Imine Thanh N. Truong Henry Eyring Center for Theoretical Chemistry, Department of Chemistry, UniVersity of Utah, Salt Lake City, Utah 84112 ReceiVed: March 25, 1998; In Final Form: July 17, 1998 The effects of aqueous solvent on structures and mechanism of the [2 + 2] cycloaddition between ketene and imine were investigated by using correlated MP2 and MP4 levels of ab initio molecular orbital theory in conjunction with the dielectric continuum Generalized Conductor-like Screening Model (GCOSMO) for solvation. We found that reactions in the gas phase and in aqueous solution have very different topology on the free energy surfaces but have similar characteristic motion along the reaction coordinate. First, it involves formation of a planar trans-conformation zwitterionic complex, then a rotation of the two moieties to form the cycloaddition product. Aqueous solvent significantly stabilizes the zwitterionic complex, consequently changing the topology of the free energy surface from a gas-phase single barrier (one-step) process to a double barrier (two-step) one with a stable intermediate. Electrostatic solvent-solute interaction was found to be the dominant factor in lowering the activation energy by 4.5 kcal/mol. The present calculated results are consistent with previous experimental data. Introduction Lim and Jorgensen have also carried out free energy perturbation (FEP) theory simulations with accurate force field that includes + [2 2] cycloaddition reactions are useful synthetic routes to solute polarization effects and found even much larger solvent - formation of four-membered rings. -

Polyphenolic Compounds Extracted and Purified from Buddleja Globosa
molecules Article Polyphenolic Compounds Extracted and Purified from Buddleja Globosa Hope (Buddlejaceae) Leaves Using Natural Deep Eutectic Solvents and Centrifugal Partition Chromatography Jeniffer Torres-Vega 1 , Sergio Gómez-Alonso 2 , José Pérez-Navarro 2 , Julio Alarcón-Enos 3 and Edgar Pastene-Navarrete 1,3,* 1 Laboratorio de Farmacognosia, Departamento de Farmacia, Facultad de Farmacia, Universidad de Concepción, Concepción PC4030000, Chile; [email protected] 2 Regional Institute for Applied Scientific Research, Faculty of Chemical Sciences, University of Castilla-La Mancha, PC13071 Castilla-La Mancha, Spain; [email protected] (S.G.-A.); [email protected] (J.P.-N.) 3 Laboratorio de Síntesis y Biotransformación de Productos Naturales, Universidad del Bío-Bío, Chillán PC3800708, Chile; [email protected] * Correspondence: [email protected]; Tel.: +56-(42)-246-3000 Abstract: Chemical profiling of Buddleja globosa was performed by high-performance liquid chro- matography coupled to electrospray ionization (HPLC-DAD-ESI-IT/MS) and quadrupole time-of-flight high-resolution mass spectrometry (HPLC-ESI-QTOF/MS). The identification of 17 main phenolic com- pounds in B. globosa leaf extracts was achieved. Along with caffeoyl glucoside isomers, caffeoylshikimic Citation: Torres-Vega, J.; acid and several verbascoside derivatives (β-hydroxyverbascoside and β-hydroxyisoverbascoside) were Gómez-Alonso, S.; Pérez-Navarro, J.; Alarcón-Enos, J.; Pastene-Navarrete, identified. Among flavonoid compounds, the presence of 6-hydroxyluteolin-7-O-glucoside, quercetin-3- E. Polyphenolic Compounds O-glucoside, luteolin 7-O-glucoside, apigenin 7-O-glucoside was confirmed. Campneoside I, forsytho- Extracted and Purified from Buddleja side B, lipedoside A and forsythoside A were identified along with verbascoside, isoverbascoside, Globosa Hope (Buddlejaceae) Leaves eukovoside and martynoside. -

Electron Ionization
Chapter 6 Chapter 6 Electron Ionization I. Introduction ......................................................................................................317 II. Ionization Process............................................................................................317 III. Strategy for Data Interpretation......................................................................321 1. Assumptions 2. The Ionization Process IV. Types of Fragmentation Pathways.................................................................328 1. Sigma-Bond Cleavage 2. Homolytic or Radical-Site-Driven Cleavage 3. Heterolytic or Charge-Site-Driven Cleavage 4. Rearrangements A. Hydrogen-Shift Rearrangements B. Hydride-Shift Rearrangements V. Representative Fragmentations (Spectra) of Classes of Compounds.......... 344 1. Hydrocarbons A. Saturated Hydrocarbons 1) Straight-Chain Hydrocarbons 2) Branched Hydrocarbons 3) Cyclic Hydrocarbons B. Unsaturated C. Aromatic 2. Alkyl Halides 3. Oxygen-Containing Compounds A. Aliphatic Alcohols B. Aliphatic Ethers C. Aromatic Alcohols D. Cyclic Ethers E. Ketones and Aldehydes F. Aliphatic Acids and Esters G. Aromatic Acids and Esters 4. Nitrogen-Containing Compounds A. Aliphatic Amines B. Aromatic Compounds Containing Atoms of Nitrogen C. Heterocyclic Nitrogen-Containing Compounds D. Nitro Compounds E. Concluding Remarks on the Mass Spectra of Nitrogen-Containing Compounds 5. Multiple Heteroatoms or Heteroatoms and a Double Bond 6. Trimethylsilyl Derivative 7. Determining the Location of Double Bonds VI. Library -

The Arene–Alkene Photocycloaddition
The arene–alkene photocycloaddition Ursula Streit and Christian G. Bochet* Review Open Access Address: Beilstein J. Org. Chem. 2011, 7, 525–542. Department of Chemistry, University of Fribourg, Chemin du Musée 9, doi:10.3762/bjoc.7.61 CH-1700 Fribourg, Switzerland Received: 07 January 2011 Email: Accepted: 23 March 2011 Ursula Streit - [email protected]; Christian G. Bochet* - Published: 28 April 2011 [email protected] This article is part of the Thematic Series "Photocycloadditions and * Corresponding author photorearrangements". Keywords: Guest Editor: A. G. Griesbeck benzene derivatives; cycloadditions; Diels–Alder; photochemistry © 2011 Streit and Bochet; licensee Beilstein-Institut. License and terms: see end of document. Abstract In the presence of an alkene, three different modes of photocycloaddition with benzene derivatives can occur; the [2 + 2] or ortho, the [3 + 2] or meta, and the [4 + 2] or para photocycloaddition. This short review aims to demonstrate the synthetic power of these photocycloadditions. Introduction Photocycloadditions occur in a variety of modes [1]. The best In the presence of an alkene, three different modes of photo- known representatives are undoubtedly the [2 + 2] photocyclo- cycloaddition with benzene derivatives can occur, viz. the addition, forming either cyclobutanes or four-membered hetero- [2 + 2] or ortho, the [3 + 2] or meta, and the [4 + 2] or para cycles (as in the Paternò–Büchi reaction), whilst excited-state photocycloaddition (Scheme 2). The descriptors ortho, meta [4 + 4] cycloadditions can also occur to afford cyclooctadiene and para only indicate the connectivity to the aromatic ring, and compounds. On the other hand, the well-known thermal [4 + 2] do not have any implication with regard to the reaction mecha- cycloaddition (Diels–Alder reaction) is only very rarely nism. -
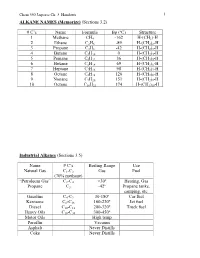
C's Name Formula Bp (ºC) Structure 1 Methane CH4 -162 H-(CH2)
Chem 350 Jasperse Ch. 3 Handouts 1 ALKANE NAMES (Memorize) (Sections 3.2) # C’s Name Formula Bp (ºC) Structure 1 Methane CH4 -162 H-(CH2)-H 2 Ethane C2H6 -89 H-(CH2)2-H 3 Propane C3H8 -42 H-(CH2)3-H 4 Butane C4H10 0 H-(CH2)4-H 5 Pentane C5H12 36 H-(CH2)5-H 6 Hexane C6H14 69 H-(CH2)6-H 7 Heptane C7H16 98 H-(CH2)7-H 8 Octane C8H18 126 H-(CH2)8-H 9 Nonane C9H20 151 H-(CH2)9-H 10 Octane C10H22 174 H-(CH2)10-H Industrial Alkanes (Sections 3.5) Name # C’s Boiling Range Use Natural Gas C1-C3 Gas Fuel (70% methane) “Petroleum Gas” C2-C4 <30º Heating, Gas Propane C3 -42º Propane tanks, camping, etc. Gasoline C4-C9 30-180º Car fuel Kerosene C8-C16 160-230º Jet fuel Diesel C10-C18 200-320º Truck fuel Heavy Oils C16-C30 300-450º Motor Oils High temp Paraffin Vacuum Asphalt Never Distills Coke Never Distills Chem 350 Jasperse Ch. 3 Handouts 2 Nomenclature of Alkanes (Sections 3.3) Systematic IUPAC Rules for Branched and Substituted Alkanes (Section 3.3B) 1. Longest continuous C-chain “core name” 2. Number core chain from an end nearest a substituent 3. Name substituents as “alkyl” groups: 4. Specify the location of substituents using numbers (hyphenate the #’s) • If >2 substituents, list alphabetically • Use di-, tri-, tetra- if the same substituent is repeated. (But ignore these in alphabetizing). Punctuation Notes: • Hyphenate numbers • Do not put a space between substituents and the core name Special Names for Some 3 or 4-carbon Substituents H3C CH3 CH Memorize H3C C H3C CH3 Isopropyl t-butyl or tert-butyl H2 H2 CH3 H3C C H3C C CH H3C CH Others C C C C CH3 H3C C H2 H H H2 2 2 H2 n-propyl n-butyl isobutyl s-butyl (n for "normal") Another Classification System Primary (1º): with one attached carbon Secondary (2º): with two attached carbons Tertiary (3º): with three attached carbons H C C C C C C C C H 1º H 2º C 3º Very Complex Substituents (Not responsible) Substituent: (1-ethyl-2,3-dimethylpentyl) Overall: 9-(1-ethyl-2,3-dimethylpentyl)nonadecane Chem 350 Jasperse Ch. -

Alkenes and Alkynes
02/21/2019 CHAPTER FOUR Alkenes and Alkynes H N O I Cl C O C O Cl F3C C Cl C Cl Efavirenz Haloprogin (antiviral, AIDS therapeutic) (antifungal, antiseptic) Chapter 4 Table of Content * Unsaturated Hydrocarbons * Introduction and hybridization * Alkenes and Alkynes * Benzene and Phenyl groups * Structure of Alkenes, cis‐trans Isomerism * Nomenclature of Alkenes and Alkynes * Configuration cis/trans, and cis/trans Isomerism * Configuration E/Z * Physical Properties of Hydrocarbons * Acid‐Base Reactions of Hydrocarbons * pka and Hybridizations 1 02/21/2019 Unsaturated Hydrocarbons • Unsaturated Hydrocarbon: A hydrocarbon that contains one or more carbon‐carbon double or triple bonds or benzene‐like rings. – Alkene: contains a carbon‐carbon double bond and has the general formula CnH2n. – Alkyne: contains a carbon‐carbon triple bond and has the general formula CnH2n‐2. Introduction Alkenes ● Hydrocarbons containing C=C ● Old name: olefins • Steroids • Hormones • Biochemical regulators 2 02/21/2019 • Alkynes – Hydrocarbons containing C≡C – Common name: acetylenes Unsaturated Hydrocarbons • Arene: benzene and its derivatives (Ch 9) 3 02/21/2019 Benzene and Phenyl Groups • We do not study benzene and its derivatives until Chapter 9. – However, we show structural formulas of compounds containing a phenyl group before that time. – The phenyl group is not reactive under any of the conditions we describe in chapters 5‐8. Structure of Alkenes • The two carbon atoms of a double bond and the four atoms bonded to them lie in a plane, with bond angles of approximately 120°. 4 02/21/2019 Structure of Alkenes • Figure 4.1 According to the orbital overlap model, a double bond consists of one bond formed by overlap of sp2 hybrid orbitals and one bond formed by overlap of parallel 2p orbitals. -

Influence of Conjugation Axis on the Optical and Electronic Properties Of
Article pubs.acs.org/joc Influence of Conjugation Axis on the Optical and Electronic Properties of Aryl-Substituted Benzobisoxazoles † ‡ ‡ † † Brian C. Tlach, Aimeé L. Tomlinson, Alden G. Ryno, Dawn D. Knoble, Dana L. Drochner, † Kyle J. Krager, and Malika Jeffries-EL*, † Department of Chemistry, Iowa State University, Ames, Iowa 50010, United States ‡ Department of Chemistry, University of North Georgia, Dahlonega, Georgia 30597, United States *S Supporting Information ABSTRACT: Six different 2,6-diethyl-4,8-diarylbenzo[1,2-d:4,5-d′]bis(oxazoles) and four different 2,4,6,8-tetraarylbenzobisoxazoles were synthesized in two steps: a Lewis acid catalyzed orthoester cyclization followed by a Suzuki or Stille cross-coupling with various arenes. The influence of aryl group substitution and/or conjugation axis variation on the optical and electronic properties of these benzobis(oxazole) (BBO) compounds was evaluated. Structural modifications could be used to alter the HOMO, LUMO, and band gap over a range of 1.0, 0.5, and 0.5 eV, respectively. However, depending on the location and identity of the substituent, the HOMO level can be altered without significantly impacting the LUMO level. This is supported by the calculated frontier molecular orbitals. Our results indicate that the FMOs and band gaps of benzobisoxazoles can be readily modified either jointly or individually. ■ INTRODUCTION Among the aforementioned examples, the benzo[1,2-d:4,5- d′]bis(oxazole) (BBO)-based cruciforms are particularly During the past four decades, interest in the development of π- interesting, since these molecules have two different con- conjugated materials has increased due to their potential use as jugation pathways: 2,6-conjugation through the oxazole rings replacements for inorganic materials in a variety of semi- and 4,8-conjugation through the central benzene ring (Scheme conducting applications including field effect transistors − − 1). -

Reactions of Aromatic Compounds Just Like an Alkene, Benzene Has Clouds of Electrons Above and Below Its Sigma Bond Framework
Reactions of Aromatic Compounds Just like an alkene, benzene has clouds of electrons above and below its sigma bond framework. Although the electrons are in a stable aromatic system, they are still available for reaction with strong electrophiles. This generates a carbocation which is resonance stabilized (but not aromatic). This cation is called a sigma complex because the electrophile is joined to the benzene ring through a new sigma bond. The sigma complex (also called an arenium ion) is not aromatic since it contains an sp3 carbon (which disrupts the required loop of p orbitals). Ch17 Reactions of Aromatic Compounds (landscape).docx Page1 The loss of aromaticity required to form the sigma complex explains the highly endothermic nature of the first step. (That is why we require strong electrophiles for reaction). The sigma complex wishes to regain its aromaticity, and it may do so by either a reversal of the first step (i.e. regenerate the starting material) or by loss of the proton on the sp3 carbon (leading to a substitution product). When a reaction proceeds this way, it is electrophilic aromatic substitution. There are a wide variety of electrophiles that can be introduced into a benzene ring in this way, and so electrophilic aromatic substitution is a very important method for the synthesis of substituted aromatic compounds. Ch17 Reactions of Aromatic Compounds (landscape).docx Page2 Bromination of Benzene Bromination follows the same general mechanism for the electrophilic aromatic substitution (EAS). Bromine itself is not electrophilic enough to react with benzene. But the addition of a strong Lewis acid (electron pair acceptor), such as FeBr3, catalyses the reaction, and leads to the substitution product. -

Predictive Models for Kinetic Parameters of Cycloaddition
Predictive Models for Kinetic Parameters of Cycloaddition Reactions Marta Glavatskikh, Timur Madzhidov, Dragos Horvath, Ramil Nugmanov, Timur Gimadiev, Daria Malakhova, Gilles Marcou, Alexandre Varnek To cite this version: Marta Glavatskikh, Timur Madzhidov, Dragos Horvath, Ramil Nugmanov, Timur Gimadiev, et al.. Predictive Models for Kinetic Parameters of Cycloaddition Reactions. Molecular Informatics, Wiley- VCH, 2018, 38 (1-2), pp.1800077. 10.1002/minf.201800077. hal-02346844 HAL Id: hal-02346844 https://hal.archives-ouvertes.fr/hal-02346844 Submitted on 5 Feb 2021 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. 0DQXVFULSW &OLFNKHUHWRGRZQORDG0DQXVFULSW&$UHYLVLRQGRF[ Predictive models for kinetic parameters of cycloaddition reactions Marta Glavatskikh[a,b], Timur Madzhidov[b], Dragos Horvath[a], Ramil Nugmanov[b], Timur Gimadiev[a,b], Daria Malakhova[b], Gilles Marcou[a] and Alexandre Varnek*[a] [a] Laboratoire de Chémoinformatique, UMR 7140 CNRS, Université de Strasbourg, 1, rue Blaise Pascal, 67000 Strasbourg, France; [b] Laboratory of Chemoinformatics and Molecular Modeling, Butlerov Institute of Chemistry, Kazan Federal University, Kremlyovskaya str. 18, Kazan, Russia Abstract This paper reports SVR (Support Vector Regression) and GTM (Generative Topographic Mapping) modeling of three kinetic properties of cycloaddition reactions: rate constant (logk), activation energy (Ea) and pre-exponential factor (logA).