Modafinil Tablets
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
![[123I]FP-CIT SPECT in Atypical Degenerative Parkinsonism](https://docslib.b-cdn.net/cover/2351/123i-fp-cit-spect-in-atypical-degenerative-parkinsonism-242351.webp)
[123I]FP-CIT SPECT in Atypical Degenerative Parkinsonism
CONTRAST AGENT EVALUATION [123I]FP-CIT SPECT in atypical degenerative parkinsonism One of the most widely used techniques to support the clinical diagnosis of Parkinson’s disease is the SPECT scan with [123I]FP-CIT. This tracer binds reversibly and visualizes the striatal presynaptic dopamine transporters. Several uncertainties remain on the value of [123I]FP-CIT and SPECT in atypical degenerative parkinsonian syndromes. In this concise review, we discuss the contribution of SPECT and [123I]FP-CIT in supporting the clinical diagnosis of Parkinson’s disease and their role in the differential diagnosis of Parkinson’s disease and atypical degenerative parkinsonism. The chemistry, pharmacodynamics and pharmacokinetics of [123I]FP-CIT are also discussed. 1,2,3 KEywordS: atypical degenerative parkinsonism n FP-CIT n ioflupane n SPECT Ioannis U Isaias* , Giorgio Marotta4, Gianni Pezzoli2, Parkinson’s disease (PD) is the second most dystonic tremor [15] and psychogenic parkin- Osama Sabri5 [1] [16,17] 5,6 common neurodegenerative disorder , yet sonism . In this concise review, we will & Swen Hesse 123 early accurate diagnosis remains challenging. discuss the role of SPECT and [ I]FP-CIT in 1Università degli Studi di Milano, The estimated prevalence of PD is 0.5–1% in supporting the clinical diagnosis of PD and its Dipartimento di Fisiologia Umana, those aged 65–69 years and 1–3% in those aged differential diagnosis with ADP. Milano, Italy 2Centro per la Malattia di Parkinson e i ≥80 years [1]. Although the clinical diagnosis of Disturbi del Movimento, -

Modafinil and Modafinil Analogues: Free Radical Mechanism of the Eugeroic and Cognitive Enhancment Effect Clifford Fong
Modafinil and modafinil analogues: free radical mechanism of the eugeroic and cognitive enhancment effect Clifford Fong To cite this version: Clifford Fong. Modafinil and modafinil analogues: free radical mechanism of the eugeroic and cognitive enhancment effect. [Research Report] Eigenenergy. 2018. hal-01933737 HAL Id: hal-01933737 https://hal.archives-ouvertes.fr/hal-01933737 Submitted on 24 Nov 2018 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Modafinil and modafinil analogues: free radical mechanism of the eugeroic and cognitive enhancment effect Clifford W. Fong Eigenenergy, Adelaide, South Australia. Keywords: Modafinil, modafinil-like analogues, eugeroic effect, cognitive enhancement, free radicals, quantum mechanics Abbreviations Dopamine DA, dopamine transporter DAT, Dissociative electron transfer or attachment DET, Linear free energy relationship LFER, free energy of water desolvation ΔG desolv,CDS , lipophilicity free energy ΔG lipo,CDS, cavity dispersion solvent structure of the first solvation shell CDS, highest occupied molecular orbital HOMO, lowest unoccupied molecular orbital LUMO, multiple correlation coefficient R 2, the F test of significance, standards errors for the estimate (SEE) and standard errors of the variables SE(ΔG desolCDS ), SE(ΔG lipoCDS ), SE(Dipole Moment), SE (Molecular Volume), transition state TS, reactive oxygen species ROS. -

These Highlights Do Not Include All the Information Needed to Use PROVIGIL Safely and Effectively
PROVIGIL- modafinil tablet Bryant Ranch Prepack ---------- HIGHLIGHTS OF PRESCRIBING INFORMATION These highlights do not include all the information needed to use PROVIGIL safely and effectively. See full prescribing information for PROVIGIL. PROVIGIL® (modafinil) tablets, for oral use, C-IV Initial U.S. Approval: 1998 INDICATIONS AND USAGE PROVIGIL is indicated to improve wakefulness in adult patients with excessive sleepiness associated with narcolepsy, obstructive sleep apnea (OSA), or shift work disorder (SWD). (1) Limitations of Use In OSA, PROVIGIL is indicated to treat excessive sleepiness and not as treatment for the underlying obstruction. DOSAGE AND ADMINISTRATION The recommended dosage of PROVIGIL for each indication is as follows: • Narcolepsy or OSA: 200 mg once a day in the morning. (2.1) • SWD: 200 mg once a day, taken approximately one hour prior to start of the work shift. (2.2) • Severe Hepatic Impairment: reduce dose to half the recommended dose. (2.3, 12.3) • Geriatric Patients: consider lower dose. (2.4, 12.3) DOSAGE FORMS AND STRENGTHS Tablets: 100 mg and 200 mg. (3) CONTRAINDICATIONS PROVIGIL is contraindicated in patients with known hypersensitivity to modafinil or armodafinil. (4) WARNINGS AND PRECAUTIONS • Serious Rash, including Stevens-Johnson Syndrome: Discontinue PROVIGIL at the first sign of rash, unless the rash is clearly not drug-related. (5.1) • Angioedema and Anaphylaxis Reactions: If suspected, discontinue PROVIGIL. (5.2) • Multi-organ Hypersensitivity Reactions: If suspected, discontinue PROVIGIL. (5.3) • Persistent Sleepiness: Assess patients frequently for degree of sleepiness and, if appropriate, advise patients to avoid driving or engaging in any other potentially dangerous activity. (5.4) • Psychiatric Symptoms: Use caution in patients with a history of psychosis, depression, or mania. -

Contribution of Mass Spectrometry for the Detection of Xenobiotics
Contribution of mass spectrometry for the detection of xenobiotics implicated in cases of drug-facilitated crimes and the quantitation of urinary metabolites of polycyclic aromatic hydrocarbons Mohammed K.S. Shbair To cite this version: Mohammed K.S. Shbair. Contribution of mass spectrometry for the detection of xenobiotics implicated in cases of drug-facilitated crimes and the quantitation of urinary metabolites of polycyclic aromatic hydrocarbons. Human health and pathology. Université du Droit et de la Santé - Lille II, 2011. English. NNT : 2011LIL2S011. tel-00647316 HAL Id: tel-00647316 https://tel.archives-ouvertes.fr/tel-00647316 Submitted on 1 Dec 2011 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. University Lille 2 of Health and Law Doctorate School of Health and Biology Thesis for the Degree of Doctorate of Lille 2 University Discipline: TOXICOLOGY By Mohammed K.S. SHBAIR Contribution of mass spectrometry for the detection of xenobiotics implicated in cases of drug-facilitated crimes and the quantitation of urinary metabolites of polycyclic aromatic hydrocarbons Thesis defended 23rd June 2011 Members of the jury: Reviewers: Prof. Dr. Jean-Pierre GOULLÉ Prof. Dr. Christian STAUB Examiners : Prof. Dr. -

Texas Controlled Substances Act
HEALTH AND SAFETY CODE TITLE 6. FOOD, DRUGS, ALCOHOL, AND HAZARDOUS SUBSTANCES SUBTITLE C. SUBSTANCE ABUSE REGULATION AND CRIMES CHAPTER 481. TEXAS CONTROLLED SUBSTANCES ACT SUBCHAPTER A. GENERAL PROVISIONS Sec.A481.001.AASHORT TITLE. This chapter may be cited as the Texas Controlled Substances Act. Acts 1989, 71st Leg., ch. 678, Sec. 1, eff. Sept. 1, 1989. Sec.A481.002.AADEFINITIONS. In this chapter: (1)AA"Administer" means to directly apply a controlled substance by injection, inhalation, ingestion, or other means to the body of a patient or research subject by: (A)AAa practitioner or an agent of the practitioner in the presence of the practitioner; or (B)AAthe patient or research subject at the direction and in the presence of a practitioner. (2)AA"Agent" means an authorized person who acts on behalf of or at the direction of a manufacturer, distributor, or dispenser. The term does not include a common or contract carrier, public warehouseman, or employee of a carrier or warehouseman acting in the usual and lawful course of employment. (3)AA"Commissioner" means the commissioner of state health services or the commissioner 's designee. (4)AA"Controlled premises" means: (A)AAa place where original or other records or documents required under this chapter are kept or are required to be kept; or (B)AAa place, including a factory, warehouse, other establishment, or conveyance, where a person registered under this chapter may lawfully hold, manufacture, distribute, dispense, administer, possess, or otherwise dispose of a controlled substance or other item governed by the federal Controlled Substances Act (21 U.S.C. -

Pharmacy and Poisons Act 1979
Q UO N T FA R U T A F E BERMUDA PHARMACY AND POISONS ACT 1979 1979 : 26 TABLE OF CONTENTS PART I PRELIMINARY 1 Short title 2 Interpretation PART II THE PHARMACY COUNCIL 3 The Pharmacy Council 4 Membership of the Council 4A Functions of the Council 4B Protection from personal liability 4C Annual Report 5 Proceedings of the Council, etc PART III REGISTRATION OF PHARMACISTS 6 Offence to practise pharmacy if not registered 7 Registration as a pharmacist 7A Re-registration as non-practising member 7AA Period of validity of registration 8 Code of Conduct 9 Pharmacy Profession Complaints Committee 10 Investigation of complaint by Committee 10A Inquiry into complaint by Council 10B Inquiry by Council of its own initiative 11 Surrender of registration 12 Restoration of name to register 1 PHARMACY AND POISONS ACT 1979 13 Proof of registration 14 Appeals 14A Fees 14B Amendment of Seventh Schedule 15 Regulations for this part PART IV REGISTRATION OF PHARMACIES 16 Register of pharmacies 17 Registration of premises as registered pharmacies 18 Unfit premises: new applications 19 Unfit premises: registered pharmacies 20 Appeals 21 When certificates of unfitness take effect 22 Regulations for this Part PART V CONTROL OF PRESCRIPTIONS AND IMPORTATION 23 Prescriptions to be in a certain form 23A Validity of a prescription 24 Supply by registered pharmacist of equivalent medicines 25 Restrictions on the importation of medicines 26 Declaration relating to imported medicines [repealed] PART VI CONTROL OF DRUGS 27 Certain substances to be sold on prescription -

Modafinil: a Review of Neurochemical Actions and Effects on Cognition
Neuropsychopharmacology (2008) 33, 1477–1502 & 2008 Nature Publishing Group All rights reserved 0893-133X/08 $30.00 www.neuropsychopharmacology.org Perspective Modafinil: A Review of Neurochemical Actions and Effects on Cognition ,1 1 Michael J Minzenberg* and Cameron S Carter 1Imaging Research Center, Davis School of Medicine, UC-Davis Health System, University of California, Sacramento, CA, USA Modafinil (2-[(Diphenylmethyl) sulfinyl] acetamide, Provigil) is an FDA-approved medication with wake-promoting properties. Pre-clinical studies of modafinil suggest a complex profile of neurochemical and behavioral effects, distinct from those of amphetamine. In addition, modafinil shows initial promise for a variety of off-label indications in psychiatry, including treatment-resistant depression, attention- deficit/hyperactivity disorder, and schizophrenia. Cognitive dysfunction may be a particularly important emerging treatment target for modafinil, across these and other neuropsychiatric disorders. We aimed to comprehensively review the empirical literature on neurochemical actions of modafinil, and effects on cognition in animal models, healthy adult humans, and clinical populations. We searched PubMed with the search term ‘modafinil’ and reviewed all English-language articles for neurochemical, neurophysiological, cognitive, or information-processing experimental measures. We additionally summarized the pharmacokinetic profile of modafinil and clinical efficacy in psychiatric patients. Modafinil exhibits robust effects on catecholamines, serotonin, glutamate, gamma amino-butyric acid, orexin, and histamine systems in the brain. Many of these effects may be secondary to catecholamine effects, with some selectivity for cortical over subcortical sites of action. In addition, modafinil (at well-tolerated doses) improves function in several cognitive domains, including working memory and episodic memory, and other processes dependent on prefrontal cortex and cognitive control. -

3514-18 Dowvigil P-I.FH10
160 mm 01 ® of expression of CYP2C9 activity. Other CYP activities may not appear to be affected 100mg Tablets by modafinil. Dowvigil 200mg Tablets Potential Interactions with Drugs that Inhibit, Induce, or are Metabolized by (Modafinil) Cytochrome P450 Isoenzymes and Other Hepatic Enzymes There may be a low probability of substantive effects on the overall pharmacokinetic profile of modafinil due to CYP inhibition by concomitant medications. Due to the partial DESCRIPTION involvement of CYP3A enzymes in the metabolic elimination of modafinil, coadministration Dowvigil (modafinil) is a wakefulness promoting agent for oral administration. Modafinil of potent inducers of CYP3A4/5 (e.g., carbamazepine, phenobarbital, and rifampin) or is a racemic compound. The chemical name for modafinil is 2-[(diphenylmethyl) inhibitors of CYP3A4/5 (e.g., ketoconazole, erythromycin) may alter the plasma sulfinyl]acetamide. The molecular formula is C15H15NO2S and the molecular weight is concentrations of modafinil. 273.35. The Potential of Modafinil to Alter the Metabolism of Other Drugs by Enzyme Induction or Inhibition COMPOSITION Drugs Metabolized by CYP3A4/5 Each tablet contains: Modafinil is a weak inducer of CYP3A activity in a concentration-related manner. Modafinil (USP)……………………………… 100mg Therefore, the blood levels and effectiveness of drugs that are substrates for CYP3A enzymes (e.g., steroidal contraceptives, cyclosporine, midazolam, and triazolam) may Each tablet contains: be reduced after initiation of concomitant treatment with modafinil. Modafinil (USP)……...……………………… 200mg Ethinyl Estradiol: Administration of modafinil once daily at 200mg/day for 7 days followed by 400mg/day for 21 days may result in a mean 11% decrease in mean Cmax THERAPEUTIC INDICATIONS and 18% decrease in mean AUC0-24 of ethinyl estradiol.There may be no apparent Dowvigil is indicated to improve wakefulness in adult individuals with excessive change in the elimination rate of ethinyl estradiol. -
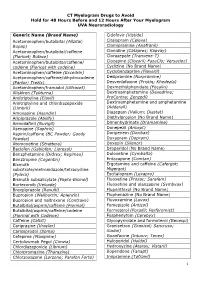
CT Myelogram Drugs to Avoid Hold for 48 Hours Before and 12 Hours After Your Myelogram UVA Neuroradiology
CT Myelogram Drugs to Avoid Hold for 48 Hours Before and 12 Hours After Your Myelogram UVA Neuroradiology Generic Name (Brand Name) Cidofovir (Vistide) Acetaminophen/butalbital (Allzital; Citalopram (Celexa) Bupap) Clomipramine (Anafranil) Acetaminophen/butalbital/caffeine Clonidine (Catapres; Kapvay) (Fioricet; Butace) Clorazepate (Tranxene-T) Acetaminophen/butalbital/caffeine/ Clozapine (Clozaril; FazaClo; Versacloz) codeine (Fioricet with codeine) Cyclizine (No Brand Name) Acetaminophen/caffeine (Excedrin) Cyclobenzaprine (Flexeril) Acetaminophen/caffeine/dihydrocodeine Desipramine (Norpramine) (Panlor; Trezix) Desvenlafaxine (Pristiq; Khedezla) Acetaminophen/tramadol (Ultracet) Dexmethylphenidate (Focalin) Aliskiren (Tekturna) Dextroamphetamine (Dexedrine; Amitriptyline (Elavil) ProCentra; Zenzedi) Amitriptyline and chlordiazepoxide Dextroamphetamine and amphetamine (Limbril) (Adderall) Amoxapine (Asendin) Diazepam (Valium; Diastat) Aripiprazole (Abilify) Diethylpropion (No Brand Name) Armodafinil (Nuvigil) Dimenhydrinate (Dramamine) Asenapine (Saphris) Donepezil (Aricept) Aspirin/caffeine (BC Powder; Goody Doripenem (Doribax) Powder) Doxapram (Dopram) Atomoxetine (Strattera) Doxepin (Silenor) Baclofen (Gablofen; Lioresal) Droperidol (No Brand Name) Benzphetamine (Didrex; Regimex) Duloxetine (Cymbalta) Benztropine (Cogentin) Entacapone (Comtan) Bismuth Ergotamine and caffeine (Cafergot; subcitrate/metronidazole/tetracycline Migergot) (Pylera) Escitalopram (Lexapro) Bismuth subsalicylate (Pepto-Bismol) Fluoxetine (Prozac; Sarafem) -

Guideline for Preoperative Medication Management
Guideline: Preoperative Medication Management Guideline for Preoperative Medication Management Purpose of Guideline: To provide guidance to physicians, advanced practice providers (APPs), pharmacists, and nurses regarding medication management in the preoperative setting. Background: Appropriate perioperative medication management is essential to ensure positive surgical outcomes and prevent medication misadventures.1 Results from a prospective analysis of 1,025 patients admitted to a general surgical unit concluded that patients on at least one medication for a chronic disease are 2.7 times more likely to experience surgical complications compared with those not taking any medications. As the aging population requires more medication use and the availability of various nonprescription medications continues to increase, so does the risk of polypharmacy and the need for perioperative medication guidance.2 There are no well-designed trials to support evidence-based recommendations for perioperative medication management; however, general principles and best practice approaches are available. General considerations for perioperative medication management include a thorough medication history, understanding of the medication pharmacokinetics and potential for withdrawal symptoms, understanding the risks associated with the surgical procedure and the risks of medication discontinuation based on the intended indication. Clinical judgement must be exercised, especially if medication pharmacokinetics are not predictable or there are significant risks associated with inappropriate medication withdrawal (eg, tolerance) or continuation (eg, postsurgical infection).2 Clinical Assessment: Prior to instructing the patient on preoperative medication management, completion of a thorough medication history is recommended – including all information on prescription medications, over-the-counter medications, “as needed” medications, vitamins, supplements, and herbal medications. Allergies should also be verified and documented. -

The Use and Impact of Cognitive Enhancers Among University Students: a Systematic Review
brain sciences Systematic Review The Use and Impact of Cognitive Enhancers among University Students: A Systematic Review Safia Sharif 1 , Amira Guirguis 1,2,* , Suzanne Fergus 1,* and Fabrizio Schifano 1 1 Psychopharmacology, Substance Misuse and Novel Psychoactive Substances Research Unit, School of Life and Medical Sciences, University of Hertfordshire, Hatfield AL10 9AB, UK; [email protected] (S.S.); [email protected] (F.S.) 2 Institute of Life Sciences 2, Swansea University, Swansea SA2 8PP, Wales, UK * Correspondence: [email protected] (A.G.); [email protected] (S.F.) Abstract: Introduction: Cognitive enhancers (CEs), also known as “smart drugs”, “study aids” or “nootropics” are a cause of concern. Recent research studies investigated the use of CEs being taken as study aids by university students. This manuscript provides an overview of popular CEs, focusing on a range of drugs/substances (e.g., prescription CEs including amphetamine salt mixtures, methylphenidate, modafinil and piracetam; and non-prescription CEs including caffeine, cobalamin (vitamin B12), guarana, pyridoxine (vitamin B6) and vinpocetine) that have emerged as being misused. The diverted non-prescription use of these molecules and the related potential for dependence and/or addiction is being reported. It has been demonstrated that healthy students (i.e., those without any diagnosed mental disorders) are increasingly using drugs such as methylphenidate, a mixture of dextroamphetamine/amphetamine, and modafinil, for the purpose of increasing their alertness, concentration or memory. Aim: To investigate the level of knowledge, perception and impact of the use of a range of CEs within Higher Education Institutions. -

Appendix-2Final.Pdf 663.7 KB
North West ‘Through the Gate Substance Misuse Services’ Drug Testing Project Appendix 2 – Analytical methodologies Overview Urine samples were analysed using three methodologies. The first methodology (General Screen) was designed to cover a wide range of analytes (drugs) and was used for all analytes other than the synthetic cannabinoid receptor agonists (SCRAs). The analyte coverage included a broad range of commonly prescribed drugs including over the counter medications, commonly misused drugs and metabolites of many of the compounds too. This approach provided a very powerful drug screening tool to investigate drug use/misuse before and whilst in prison. The second methodology (SCRA Screen) was specifically designed for SCRAs and targets only those compounds. This was a very sensitive methodology with a method capability of sub 100pg/ml for over 600 SCRAs and their metabolites. Both methodologies utilised full scan high resolution accurate mass LCMS technologies that allowed a non-targeted approach to data acquisition and the ability to retrospectively review data. The non-targeted approach to data acquisition effectively means that the analyte coverage of the data acquisition was unlimited. The only limiting factors were related to the chemical nature of the analyte being looked for. The analyte must extract in the sample preparation process; it must chromatograph and it must ionise under the conditions used by the mass spectrometer interface. The final limiting factor was presence in the data processing database. The subsequent study of negative MDT samples across the North West and London and the South East used a GCMS methodology for anabolic steroids in addition to the General and SCRA screens.