UC Merced Electronic Theses and Dissertations
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Chapter 12 Lecture Notes
Chemical Kinetics “The study of speeds of reactions and the nanoscale pathways or rearrangements by which atoms and molecules are transformed to products” Chapter 13: Chemical Kinetics: Rates of Reactions © 2008 Brooks/Cole 1 © 2008 Brooks/Cole 2 Reaction Rate Reaction Rate Combustion of Fe(s) powder: © 2008 Brooks/Cole 3 © 2008 Brooks/Cole 4 Reaction Rate Reaction Rate + Change in [reactant] or [product] per unit time. Average rate of the Cv reaction can be calculated: Time, t [Cv+] Average rate + Cresol violet (Cv ; a dye) decomposes in NaOH(aq): (s) (mol / L) (mol L-1 s-1) 0.0 5.000 x 10-5 13.2 x 10-7 10.0 3.680 x 10-5 9.70 x 10-7 20.0 2.710 x 10-5 7.20 x 10-7 30.0 1.990 x 10-5 5.30 x 10-7 40.0 1.460 x 10-5 3.82 x 10-7 + - 50.0 1.078 x 10-5 Cv (aq) + OH (aq) → CvOH(aq) 2.85 x 10-7 60.0 0.793 x 10-5 -7 + + -5 1.82 x 10 change in concentration of Cv Δ [Cv ] 80.0 0.429 x 10 rate = = 0.99 x 10-7 elapsed time Δt 100.0 0.232 x 10-5 © 2008 Brooks/Cole 5 © 2008 Brooks/Cole 6 1 Reaction Rates and Stoichiometry Reaction Rates and Stoichiometry Cv+(aq) + OH-(aq) → CvOH(aq) For any general reaction: a A + b B c C + d D Stoichiometry: The overall rate of reaction is: Loss of 1 Cv+ → Gain of 1 CvOH Rate of Cv+ loss = Rate of CvOH gain 1 Δ[A] 1 Δ[B] 1 Δ[C] 1 Δ[D] Rate = − = − = + = + Another example: a Δt b Δt c Δt d Δt 2 N2O5(g) 4 NO2(g) + O2(g) Reactants decrease with time. -

Chapter 9. Atomic Transport Properties
NEA/NSC/R(2015)5 Chapter 9. Atomic transport properties M. Freyss CEA, DEN, DEC, Centre de Cadarache, France Abstract As presented in the first chapter of this book, atomic transport properties govern a large panel of nuclear fuel properties, from its microstructure after fabrication to its behaviour under irradiation: grain growth, oxidation, fission product release, gas bubble nucleation. The modelling of the atomic transport properties is therefore the key to understanding and predicting the material behaviour under irradiation or in storage conditions. In particular, it is noteworthy that many modelling techniques within the so-called multi-scale modelling scheme of materials make use of atomic transport data as input parameters: activation energies of diffusion, diffusion coefficients, diffusion mechanisms, all of which are then required to be known accurately. Modelling approaches that are readily used or which could be used to determine atomic transport properties of nuclear materials are reviewed here. They comprise, on the one hand, static atomistic calculations, in which the migration mechanism is fixed and the corresponding migration energy barrier is calculated, and, on the other hand, molecular dynamics calculations and kinetic Monte-Carlo simulations, for which the time evolution of the system is explicitly calculated. Introduction In nuclear materials, atomic transport properties can be a combination of radiation effects (atom collisions) and thermal effects (lattice vibrations), this latter being possibly enhanced by vacancies created by radiation damage in the crystal lattice (radiation enhanced-diffusion). Thermal diffusion usually occurs at high temperature (typically above 1 000 K in nuclear ceramics), which makes the transport processes difficult to model at lower temperatures because of the low probability to see the diffusion event occur in a reasonable simulation time. -

Eyring Equation
Search Buy/Sell Used Reactors Glass microreactors Hydrogenation Reactor Buy Or Sell Used Reactors Here. Save Time Microreactors made of glass and lab High performance reactor technology Safe And Money Through IPPE! systems for chemical synthesis scale-up. Worldwide supply www.IPPE.com www.mikroglas.com www.biazzi.com Reactors & Calorimeters Induction Heating Reacting Flow Simulation Steam Calculator For Process R&D Laboratories Check Out Induction Heating Software & Mechanisms for Excel steam table add-in for water Automated & Manual Solutions From A Trusted Source. Chemical, Combustion & Materials and steam properties www.helgroup.com myewoss.biz Processes www.chemgoodies.com www.reactiondesign.com Eyring Equation Peter Keusch, University of Regensburg German version "If the Lord Almighty had consulted me before embarking upon the Creation, I should have recommended something simpler." Alphonso X, the Wise of Spain (1223-1284) "Everything should be made as simple as possible, but not simpler." Albert Einstein Both the Arrhenius and the Eyring equation describe the temperature dependence of reaction rate. Strictly speaking, the Arrhenius equation can be applied only to the kinetics of gas reactions. The Eyring equation is also used in the study of solution reactions and mixed phase reactions - all places where the simple collision model is not very helpful. The Arrhenius equation is founded on the empirical observation that rates of reactions increase with temperature. The Eyring equation is a theoretical construct, based on transition state model. The bimolecular reaction is considered by 'transition state theory'. According to the transition state model, the reactants are getting over into an unsteady intermediate state on the reaction pathway. -

Ochem ACS Review 23 Aryl Halides
ACS Review Aryl Halides 1. Which one of the following has the weakest carbon-chlorine bond? A. I B. II C. III D. IV 2. Which compound in each of the following pairs is the most reactive to the conditions indicated? A. I and III B. I and IV C. II and III D. II and IV 3. Which of the following reacts at the fastest rate with potassium methoxide (KOCH 3) in methanol? A. fluorobenzene B. 4-nitrofluorobenzene C. 2,4-dinitrofluorobenzene D. 2,4,6-trinitrofluorobenzene 4. Which of the following reacts at the fastest rate with potassium methoxide (KOCH 3) in methanol? A. fluorobenzene B. p-nitrofluorobenzene C. p-fluorotoluene D. p-bromofluorobenzene 5. Which of the following is the kinetic rate equation for the addition-elimination mechanism of nucleophilic aromatic substitution? A. rate = k[aryl halide] B. rate = k[nucleophile] C. rate = k[aryl halide][nucleophile] D. rate = k[aryl halide][nucleophile] 2 6. Which of the following is not a resonance form of the intermediate in the nucleophilic addition of hydroxide ion to para -fluoronitrobenzene? A. I B. II C. III D. IV 7. Which chlorine is most susceptible to nucleophilic substitution with NaOCH 3 in methanol? A. #1 B. #2 C. #1 and #2 are equally susceptible D. no substitution is possible 8. What is the product of the following reaction? A. N,N-dimethylaniline B. para -chloro-N,N-dimethylaniline C. phenyllithium (C 6H5Li) D. meta -chloro-N,N-dimethylaniline 9. Which one of the reagents readily reacts with bromobenzene without heating? A. -

Enzymatic Kinetic Isotope Effects from First-Principles Path Sampling
Article pubs.acs.org/JCTC Enzymatic Kinetic Isotope Effects from First-Principles Path Sampling Calculations Matthew J. Varga and Steven D. Schwartz* Department of Chemistry and Biochemistry, University of Arizona, Tucson, Arizona 85721, United States *S Supporting Information ABSTRACT: In this study, we develop and test a method to determine the rate of particle transfer and kinetic isotope effects in enzymatic reactions, specifically yeast alcohol dehydrogenase (YADH), from first-principles. Transition path sampling (TPS) and normal mode centroid dynamics (CMD) are used to simulate these enzymatic reactions without knowledge of their reaction coordinates and with the inclusion of quantum effects, such as zero-point energy and tunneling, on the transferring particle. Though previous studies have used TPS to calculate reaction rate constants in various model and real systems, it has not been applied to a system as large as YADH. The calculated primary H/D kinetic isotope effect agrees with previously reported experimental results, within experimental error. The kinetic isotope effects calculated with this method correspond to the kinetic isotope effect of the transfer event itself. The results reported here show that the kinetic isotope effects calculated from first-principles, purely for barrier passage, can be used to predict experimental kinetic isotope effects in enzymatic systems. ■ INTRODUCTION staging method from the Chandler group.10 Another method that uses an energy gap formalism like QCP is a hybrid Particle transfer plays a crucial role in a breadth of enzymatic ff reactions, including hydride, hydrogen atom, electron, and quantum-classical approach of Hammes-Schi er and co-work- 1 ers, which models the transferring particle nucleus as a proton-coupled electron transfer reactions. -

An Improved Potential Energy Surface for the H2cl System and Its Use for Calculations of Rate Coefficients and Kinetic Isotope Effects Thomas C
Chemistry Publications Chemistry 8-1996 An Improved Potential Energy Surface for the H2Cl System and Its Use for Calculations of Rate Coefficients and Kinetic Isotope Effects Thomas C. Allison University of Minnesota - Twin Cities Gillian C. Lynch University of Minnesota - Twin Cities Donald G. Truhlar University of Minnesota - Twin Cities Mark S. Gordon Iowa State University, [email protected] Follow this and additional works at: http://lib.dr.iastate.edu/chem_pubs Part of the Chemistry Commons The ompc lete bibliographic information for this item can be found at http://lib.dr.iastate.edu/ chem_pubs/327. For information on how to cite this item, please visit http://lib.dr.iastate.edu/ howtocite.html. This Article is brought to you for free and open access by the Chemistry at Iowa State University Digital Repository. It has been accepted for inclusion in Chemistry Publications by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. An Improved Potential Energy Surface for the H2Cl System and Its Use for Calculations of Rate Coefficients and Kinetic Isotope Effects Abstract We present a new potential energy surface (called G3) for the chemical reaction Cl + H2 → HCl + H. The new surface is based on a previous potential surface called GQQ, and it incorporates an improved bending potential that is fit ot the results of ab initio electronic structure calculations. Calculations based on variational transition state theory with semiclassical transmission coefficients corresponding to an optimized multidimensional tunneling treatment (VTST/OMT, in particular improved canonical variational theory with least-action ground-state transmission coefficients) are carried out for nine different isotopomeric versions of the abstraction reaction and six different isotopomeric versions of the exchange reaction involving the H, D, and T isotopes of hydrogen, and the new surface is tested by comparing these calculations to available experimental data. -

Spring 2013 Lecture 13-14
CHM333 LECTURE 13 – 14: 2/13 – 15/13 SPRING 2013 Professor Christine Hrycyna INTRODUCTION TO ENZYMES • Enzymes are usually proteins (some RNA) • In general, names end with suffix “ase” • Enzymes are catalysts – increase the rate of a reaction – not consumed by the reaction – act repeatedly to increase the rate of reactions – Enzymes are often very “specific” – promote only 1 particular reaction – Reactants also called “substrates” of enzyme catalyst rate enhancement non-enzymatic (Pd) 102-104 fold enzymatic up to 1020 fold • How much is 1020 fold? catalyst time for reaction yes 1 second no 3 x 1012 years • 3 x 1012 years is 500 times the age of the earth! Carbonic Anhydrase Tissues ! + CO2 +H2O HCO3− +H "Lungs and Kidney 107 rate enhancement Facilitates the transfer of carbon dioxide from tissues to blood and from blood to alveolar air Many enzyme names end in –ase 89 CHM333 LECTURE 13 – 14: 2/13 – 15/13 SPRING 2013 Professor Christine Hrycyna Why Enzymes? • Accelerate and control the rates of vitally important biochemical reactions • Greater reaction specificity • Milder reaction conditions • Capacity for regulation • Enzymes are the agents of metabolic function. • Metabolites have many potential pathways • Enzymes make the desired one most favorable • Enzymes are necessary for life to exist – otherwise reactions would occur too slowly for a metabolizing organis • Enzymes DO NOT change the equilibrium constant of a reaction (accelerates the rates of the forward and reverse reactions equally) • Enzymes DO NOT alter the standard free energy change, (ΔG°) of a reaction 1. ΔG° = amount of energy consumed or liberated in the reaction 2. -

S.T.E.T.Women's College, Mannargudi Semester Iii Ii M
S.T.E.T.WOMEN’S COLLEGE, MANNARGUDI SEMESTER III II M.Sc., CHEMISTRY ORGANIC CHEMISTRY - II – P16CH31 UNIT I Aliphatic nucleophilic substitution – mechanisms – SN1, SN2, SNi – ion-pair in SN1 mechanisms – neighbouring group participation, non-classical carbocations – substitutions at allylic and vinylic carbons. Reactivity – effect of structure, nucleophile, leaving group and stereochemical factors – correlation of structure with reactivity – solvent effects – rearrangements involving carbocations – Wagner-Meerwein and dienone-phenol rearrangements. Aromatic nucleophilic substitutions – SN1, SNAr, Benzyne mechanism – reactivity orientation – Ullmann, Sandmeyer and Chichibabin reaction – rearrangements involving nucleophilic substitution – Stevens – Sommelet Hauser and von-Richter rearrangements. NUCLEOPHILIC SUBSTITUTION Mechanism of Aliphatic Nucleophilic Substitution. Aliphatic nucleophilic substitution clearly involves the donation of a lone pair from the nucleophile to the tetrahedral, electrophilic carbon bonded to a halogen. For that reason, it attracts to nucleophile In organic chemistry and inorganic chemistry, nucleophilic substitution is a fundamental class of reactions in which a leaving group(nucleophile) is replaced by an electron rich compound(nucleophile). The whole molecular entity of which the electrophile and the leaving group are part is usually called the substrate. The nucleophile essentially attempts to replace the leaving group as the primary substituent in the reaction itself, as a part of another molecule. The most general form of the reaction may be given as the following: Nuc: + R-LG → R-Nuc + LG: The electron pair (:) from the nucleophile(Nuc) attacks the substrate (R-LG) forming a new 1 bond, while the leaving group (LG) departs with an electron pair. The principal product in this case is R-Nuc. The nucleophile may be electrically neutral or negatively charged, whereas the substrate is typically neutral or positively charged. -

Alkyl Halides
Alkyl Halides Alkyl halides are a class of compounds where a halogen atom or atoms are bound to an sp3 orbital of an alkyl group. CHCl3 (Chloroform: organic solvent) CF2Cl2 (Freon-12: refrigerant CFC) CF3CHClBr (Halothane: anesthetic) Halogen atoms are more electronegative than carbon atoms, and so the C-Hal bond is polarized. The C-Hal (often written C-X) bond is polarized in such a way that there is partial positive charge on the carbon and partial negative charge on the halogen. Ch06 Alkyl Halides (landscape).docx Page 1 Dipole moment Electronegativities decrease in the order of: F > Cl > Br > I Carbon-halogen bond lengths increase in the order of: C-F < C-Cl < C-Br < C-I Bond Dipole Moments decrease in the order of: C-Cl > C-F > C-Br > C-I = 1.56D 1.51D 1.48D 1.29D Typically the chemistry of alkyl halides is dominated by this effect, and usually results in the C-X bond being broken (either in a substitution or elimination process). This reactivity makes alkyl halides useful chemical reagents. Ch06 Alkyl Halides (landscape).docx Page 2 Nomenclature According to IUPAC, alkyl halides are treated as alkanes with a halogen (Halo-) substituent. The halogen prefixes are Fluoro-, Chloro-, Bromo- and Iodo-. Examples: Often compounds of CH2X2 type are called methylene halides. (CH2Cl2 is methylene chloride). CHX3 type compounds are called haloforms. (CHI3 is iodoform). CX4 type compounds are called carbon tetrahalides. (CF4 is carbon tetrafluoride). Alkyl halides can be primary (1°), secondary (2°) or tertiary (3°). Other types: A geminal (gem) dihalide has two halogens on the same carbon. -

Wigner's Dynamical Transition State Theory in Phase Space
Wigner’s Dynamical Transition State Theory in Phase Space: Classical and Quantum Holger Waalkens1,2, Roman Schubert1, and Stephen Wiggins1 October 15, 2018 1 School of Mathematics, University of Bristol, University Walk, Bristol BS8 1TW, UK 2 Department of Mathematics, University of Groningen, Nijenborgh 9, 9747 AG Groningen, The Netherlands E-mail: [email protected], [email protected], [email protected] Abstract We develop Wigner’s approach to a dynamical transition state theory in phase space in both the classical and quantum mechanical settings. The key to our de- velopment is the construction of a normal form for describing the dynamics in the neighborhood of a specific type of saddle point that governs the evolution from re- actants to products in high dimensional systems. In the classical case this is the standard Poincar´e-Birkhoff normal form. In the quantum case we develop a normal form based on the Weyl calculus and an explicit algorithm for computing this quan- tum normal form. The classical normal form allows us to discover and compute the phase space structures that govern classical reaction dynamics. From this knowledge we are able to provide a direct construction of an energy dependent dividing sur- face in phase space having the properties that trajectories do not locally “re-cross” the surface and the directional flux across the surface is minimal. Using this, we are able to give a formula for the directional flux through the dividing surface that goes beyond the harmonic approximation. We relate this construction to the flux- flux autocorrelation function which is a standard ingredient in the expression for the reaction rate in the chemistry community. -

Lecture 17 Transition State Theory Reactions in Solution
Lecture 17 Transition State Theory Reactions in solution kd k1 A + B = AB → PK = kd/kd’ kd’ d[AB]/dt = kd[A][B] – kd’[AB] – k1[AB] [AB] = kd[A][B]/k1+kd’ d[P]/dt = k1[AB] = k2[A][B] k2 = k1kd/(k1+kd’) k2 is the effective bimolecular rate constant kd’<<k1, k2 = k1kd/k1 = kd diffusion controlled k1<<kd’, k2 = k1kd/kd’ = Kk1 activation controlled Other names: Activated complex theory and Absolute rate theory Drawbacks of collision theory: Difficult to calculate the steric factor from molecular geometry for complex molecules. The theory is applicable essentially to gaseous reactions Consider A + B → P or A + BC = AB + C k2 A + B = [AB] ╪ → P A and B form an activated complex and are in equilibrium with it. The reactions proceed through an activated or transition state which has energy higher than the reactions or the products. Activated complex Transition state • Reactants Has someone seen the Potential energy transition state? Products Reaction coordinate The rate depends on two factors, (i). Concentration of [AB] (ii). The rate at which activated complex is decomposed. ∴ Rate of reaction = [AB╪] x frequency of decomposition of AB╪ ╪ ╪ K eq = [AB ] / [A] [B] ╪ ╪ [AB ] = K eq [A] [B] The activated complex is an aggregate of atoms and assumed to be an ordinary molecule. It breaks up into products on a special vibration, along which it is unstable. The frequency of such a vibration is equal to the rate at which activated complex decompose. -d[A]/dt = -d[B]/dt = k2[A][B] Rate of reaction = [AB╪] υ ╪ = K eq υ [A] [B] Activated complex is an unstable species and is held together by loose bonds. -
Microtest III Print File
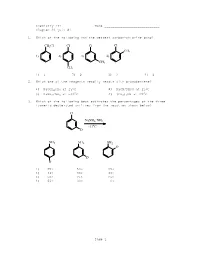
Chemistry 211 Name ____________________________ Chapter 23 Quiz #1 1. Which of the following has the weakest carbon-chlorine bond? CH2Cl Cl Cl Cl CH3 1) 2) 3) 4) CH3 CH3 1) 1 2) 2 3) 3 4) 4 2. Which one of the reagents readily reacts with bromobenzene? 1) NaOCH2CH3 at 250C 2) NaCN/DMSO at 250C 3) NaNH2/NH3 at -330C 4) (CH3)2NH at 250C 3. Which of the following best estimates the percentages of the three isomeric deuterated anilines from the reaction shown below? Cl NaNH2, NH3 -33oC D NH 2 NH 2 NH 2 D D D 1) 25% 50% 25% 2) 33% 33% 33% 3) 50% 25% 25% 4) 66% 33% 0% Page 1 4. Which of the following is (are) true concerning the intermediate in the addition-elimination mechanism of the reaction below? F OCH3 NaOCH 3 intermediate NO2 NO2 A. The intermediate is aromatic. B. The intermediate is a resonance stabilized anion. C. Electron withdrawing groups on the benzene ring stabilize the intermediate. 1) only A 2) only B 3) A and C 4) B and C 5. Carbon-14 labelled chlorobenzene is reacted with sodium amide in ammonia as shown below. Which of the following depicts the carbon-14 label in the product(s)? Cl NaNH2/NH3 -33oC Cl Cl Cl 1) 3) and (about 50% of each) Cl Cl Cl 2) 4) and (about 50% of each) 1) 1 2) 2 3) 3 4) 4 6. Which of the following reacts at the fastest rate with potassium methoxide (KOCH3) in methanol? 1) fluorobenzene 2) 4-nitrofluorobenzene 3) 2,4-dinitrofluorobenzene 4) 2,4,6-trinitrofluorobenzene Page 2 7.