De Novo Transcriptome Sequencing Coupled with Co-Expression
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Experimental Investigation of Entropic Uncertainty Relations and Coherence Uncertainty Relations
Experimental investigation of entropic uncertainty relations and coherence uncertainty relations Zhi-Yong Ding,1, 2, 3 Huan Yang,1, 4, 5 Dong Wang,1, 6, ∗ Hao Yuan,1, 6, 7 Jie Yang,1 and Liu Ye1, y 1School of Physics and Material Science, Anhui University, Hefei 230601, China 2School of Physics and Electronic Engineering, Fuyang Normal University, Fuyang 236037, China 3Key Laboratory of Functional Materials and Devices for Informatics of Anhui Educational Institutions, Fuyang Normal University, Fuyang 236037, China 4Institutes of Physical Science and Information Technology, Anhui University, Hefei 230601, China 5Department of Experiment and Practical Training Management, West Anhui University, Lu’an 237012, China 6CAS Key Laboratory of Quantum Information, University of Science and Technology of China, Hefei 230026, China 7Key Laboratory of Opto-Electronic Information Acquisition and Manipulation of Ministry of Education, Anhui University, Hefei 230601, China (Dated: December 19, 2019) Uncertainty relation usually is one of the most important features in quantum mechanics, and is the back- bone of quantum theory, which distinguishes from the rule in classical counterpart. Specifically, entropy-based uncertainty relations are of fundamental importance in the region of quantum information theory, offering one nontrivial bound of key rate towards quantum key distribution. In this work, we experimentally demonstrate the entropic uncertainty relations and coherence-based uncertainty relations in an all-optics platform. By means of preparing two kinds of bipartite initial states with high fidelity, i.e., Bell-like states and Bell-like diagonal states, we carry on local projective measurements over a complete set of mutually unbiased bases on the measured subsystem. -
![Arxiv:1909.09327V1 [Quant-Ph] 20 Sep 2019 Systems [12]](https://docslib.b-cdn.net/cover/2612/arxiv-1909-09327v1-quant-ph-20-sep-2019-systems-12-682612.webp)
Arxiv:1909.09327V1 [Quant-Ph] 20 Sep 2019 Systems [12]
Experimental certification of steering criterion based on general entropic uncertainty relation Huan Yang,1, 2, 3 Zhi-Yong Ding,1, 4, 5 Dong Wang,1, 6 Hao Yuan,1, 6, 7 Xue-Ke Song,1 Jie Yang,1 Chang-Jin Zhang,2 and Liu Ye1, ∗ 1School of Physics and Material Science, Anhui University, Hefei 230601, China 2Institutes of Physical Science and Information Technology, Anhui University, Hefei 230601, China 3Department of Experiment and Practical Training Management, West Anhui University, Luan 237012, China 4School of Physics and Electronic Engineering, Fuyang Normal University, Fuyang 236037, China 5Key Laboratory of Functional Materials and Devices for Informatics of Anhui Educational Institutions, Fuyang Normal University, Fuyang 236037, China 6CAS Key Laboratory of Quantum Information, University of Science and Technology of China, Hefei 230026, China 7Key Laboratory of Opto-Electronic Information Acquisition and Manipulation of Ministry of Education, Anhui University, Hefei 230601, China Quantum steering describes the phenomenon that one system can be immediately influenced by another with local measurements. It can be detected by the violation of a powerful and useful steering criterion from general entropic uncertainty relation. This criterion, in principle, can be evaluated straightforwardly and achieved by only probability distributions from a finite set of measurement settings. Herein, we experimentally verify the steering criterion by means of the two-photon Werner-like states and three Pauli measurements. The results indicate that quantum steering can be verified by the criterion in a convenient way. In particular, it is no need to perform the usual quantum state tomography in experiment, which reduces the required experimental resources greatly. -

The Practice and Exploration of College Physics Flipped Classroom Teaching Method
Advances in Economics, Business and Management Research, volume 75 8th International Conference on Education and Management (ICEM 2018) The Practice and Exploration of College Physics Flipped Classroom Teaching Method 1st Jian Zhang 2nd Li Wang Aviation Basic College, Aviation University of Air Force Aviation Basic College, Aviation University of Air Force Changchun, China,130022 Changchun, China,130022 [email protected] [email protected] th 3rd Bo Jiang 4 Hongliang Zhao Aviation Basic College, Aviation University of Air Force Aviation Basic College, Aviation University of Air Force Changchun, China,130022 Changchun, China,130022 [email protected] [email protected] Abstract—This paper introduces the implementation process of college physics flipped classroom teaching method, elaborates the teaching effect of teaching practice in recent years, and puts forward some suggestions for implementing this method in college physics teaching. Keywords—College Physics, Flipped Classroom, Teaching method I. INTRODUCTION The flipped classroom teaching method is emerging in recent years, make full use of the new teaching model of online teaching platform, pre-class teacher recorded a good video, together with other forms of course information uploaded to the network teaching platform, so that students in extracurricular video Learning so that students who are absent from class or who do not listen attentively to the class can keep up with the class process, which is the interaction time between the student and the teacher, the student completing the homework in class, and discussing homework with the teacher As well as points of knowledge that are not understood in the video, and teachers are used during class time to actively engage students in the learning process and provide personalized guidance [1]. -

Ethnobotanical Knowledge of Apiaceae Family in Iran: a Review
Review Article Ethnobotanical knowledge of Apiaceae family in Iran: A review Mohammad Sadegh Amiri1*, Mohammad Reza Joharchi2 1Department of Biology, Payame Noor University, Tehran, Iran 2Department of Botany, Research Center for Plant Sciences, Ferdowsi University of Mashhad, Mashhad, Iran Article history: Abstract Received: Dec 28, 2015 Objective: Apiaceae (Umbelliferae) family is one of the biggest Received in revised form: Jan 08, 2016 plant families on the earth. Iran has a huge diversity of Apiaceae Accepted: Jan10, 2016 members. This family possesses a range of compounds that have Vol. 6, No. 6, Nov-Dec 2016, many biological activities. The members of this family are well 621-635. known as vegetables, culinary and medicinal plants. Here, we present a review of ethnobotanical uses of Apiaceae plants by the * Corresponding Author: Iranian people in order to provide a comprehensive documentation Tel: +989158147889 for future investigations. Fax: +985146229291 Materials and Methods: We checked scientific studies published [email protected] in books and journals in various electronic databases (Medline, PubMed, Science Direct, Scopus and Google Scholar websites) Keywords: Apiaceae from 1937 to 2015 and reviewed a total of 52 publications that Ethnobotany provided information about different applications of these plant Medicinal Plants species in human and livestock. Non- Medicinal Plants Results: As a result of this review, several ethnobotanical usages Iran of 70 taxa, 17 of which were endemic, have been determined. These plants were used for medicinal and non-medicinal purposes. The most commonly used parts were fruits, leaves, aerial parts and gums. The most common methods of preparation were decoction, infusion and poultice. -

Identification and Functional Characterization of the First Two
Identification and functional characterization of the first two aromatic prenyltransferases implicated in the biosynthesis of furanocoumarins and prenylated coumarins in two plant families: Rutaceae and Apiaceae Fazeelat Karamat To cite this version: Fazeelat Karamat. Identification and functional characterization of the first two aromatic prenyl- transferases implicated in the biosynthesis of furanocoumarins and prenylated coumarins in two plant families: Rutaceae and Apiaceae. Agronomy. Université de Lorraine, 2013. English. NNT : 2013LORR0029. tel-01749560 HAL Id: tel-01749560 https://hal.univ-lorraine.fr/tel-01749560 Submitted on 29 Mar 2018 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. AVERTISSEMENT Ce document est le fruit d'un long travail approuvé par le jury de soutenance et mis à disposition de l'ensemble de la communauté universitaire élargie. Il est soumis à la propriété intellectuelle de l'auteur. Ceci implique une obligation de citation et de référencement lors de l’utilisation de ce document. D'autre part, toute contrefaçon, plagiat, -

Yu Guangzhong Sulla Traduzione. Proposta Di Traduzione E Commento Traduttologico Di Alcuni Saggi Critici
Corso di Laurea magistrale (ordinamento ex D.M. 270/2004) in Interpretariato e Traduzione Editoriale, Settoriale Tesi di Laurea Yu Guangzhong sulla traduzione. Proposta di traduzione e commento traduttologico di alcuni saggi critici. Relatore Dott. Paolo Magagnin Laureanda Chaonan Zang Matricola 835354 Anno Accademico 2012 / 2013 1 Alla mia famiglia e a Jian 2 摘要 余光中先生是海峡两岸知名的诗人、作家和学者,中文造诣颇高,且精通外 文;此外,他还是一位非常优秀的翻译家和翻译教育者,译作丰富,颇有研究。 本文涉及他对翻译研究的诸多思考,从《余光中谈翻译》中选取三篇具有代表性 的批评散文翻译为意大利语,并从语言学、翻译研究和文化研究的角度对翻译策 略的选择做出了详尽的分析。 本文分为四部分,第一部分是对作者生平、散文创作思想和翻译思想的简要 介绍;第二部分是三篇批评散文的意大利语译文;第三部分是本文的核心,对翻 译过程中的难点和相关翻译策略的选择做出了详尽的分析;最后一部分为术语表 和论文写作过程中使用的参考书目。 本文旨在以中文语言的独特视角,推介中国学者在翻译研究方面的看法,引 发西方读者对中国翻译研究现状的关注;同时,加强自身翻译理论修养,提高翻 译实践水平。 3 Abstract Yu Guangzhong is a famous poet, writer and scholar known in China Continental and Taiwan, who is excellent in Chinese language as well as the foreign languages e and literature. Besides, he is also an outstanding translator and educator of the translation courses, having abundant works of translation and dedicating to the translation studies. This thesis focuses on his reflection to translation studies, including the translation of three articles from his work Yu Guangzhong Talks about Translation, and a detailed analysis on the translation strategies used from the aspect of linguistics, translation studies and cultural studies. The thesis is divided into four parts. The first part is a brief introduction of Yu’s life, and a general reflection of him on the prose writing and on the translation. The second one includes the translation of three articles. Being the most important one, the third part focuses on the analysis of the translation problems and the relative strategies used to solve them. The last part is the glossary and the bibliography. -

University of Leeds Chinese Accepted Institution List 2021
University of Leeds Chinese accepted Institution List 2021 This list applies to courses in: All Engineering and Computing courses School of Mathematics School of Education School of Politics and International Studies School of Sociology and Social Policy GPA Requirements 2:1 = 75-85% 2:2 = 70-80% Please visit https://courses.leeds.ac.uk to find out which courses require a 2:1 and a 2:2. Please note: This document is to be used as a guide only. Final decisions will be made by the University of Leeds admissions teams. -
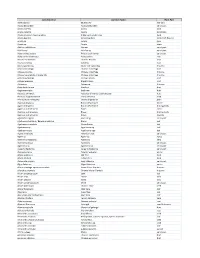
Testing Services List V2020
Latin Binomial Common Name Plant Part Abies sibirica Siberian Fir leaf (oil) Acacia Berlandieri Acacia Berlandieri aerial part Acacia catechu Acacia bark Acacia catechu Acacia gum/resin Acacia nilotica / Acacia arabica Indian gum arabic tree bark Acacia Rigidula Acacia Rigidula herb (leaf, flower) Acacia sp. Acacia gum Acacia sp. Acacia stem Achillea millefolium Yarrow aerial part Achillea sp. Achillea sp. aerial part Achyranthes aspera Prickly chaff flower aerial part Achyranthes bidentata Achyranthes root Aconite carmichaeli Chinese Aconite root Acorus calamus Calamus root Acorus gramineus Grass-leaf sweetflag rhizome Actaea cimicifuga Chinese cimicifuga root Actaea dahurica Chinese cimicifuga rhizome Actaea heracleifolia / Sheng Ma Chinese cimicifuga rhizome Actaea podocarpa Yellow Cohosh root Actaea racemosa Black Cohosh root Actaea sp. Actaea sp. rhizome Actinidia deliciosa Kiwifruit fruit Aegle marmelos Bael tree fruit Aesculus chinensis Aesculus chinensis [Sapindaceae] fruit Aesculus hippocastanum Horse chestnut seed Aframomum melegueta Grains of paradise grain Agaricus bisporus Button Mushroom entire Agaricus bisporus Button Mushroom fruiting body Agaricus subrufescens Blazei entire Agaricus subrufescens Blazei fruiting body Agaricus subrufescens Blazei mycelia Agastache rugosa Huo Xiang aerial part Agathosma betulina / Barosma betulina Buchu leaf Agathosma crenulata Ovate Buchu leaf Agathosma sp. Agathosma sp. leaf Agathosma spp. Agathosma spp. leaf Agave americana American aloe aerial part Agave sp. Agave sp. syrup Agrimonia -

16010150959.Pdf
Int. J. Electrochem. Sci., 16 (2021) 150959 International Journal of ELECTROCHEMICAL SCIENCE www.electrochemsci.org Short Communication Simulation and Experimental Research on Electrochemical Machining of Cross Groove Hua Lin1,2, Yuanlong Chen1, Xiang Li1, Huigui Li1, Qi Chen1,* 1 School of Mechanical Engineering, Hefei University of Technology, Hefei 230009, China 2 School of Mechanical and Automotive Engineering, West Anhui University, Lu’an 237000, China *E-mail: [email protected] doi: 10.20964/2021.01.20 Received: 14 September 2020 / Accepted: 27 October 2020 / Published: 30 November 2020 The cross groove and its array structure are widely used in the aviation industry and the friction surface texture. Due to its unique advantages, the electrochemical machining (ECM) has broad prospects in the processing of special-shaped holes and grooves. In this work, the planar ECM of a single cross groove was taken as the object, and a cathode with an internal waist-shaped cross groove structure was designed. Further, and a three-dimensional multi-physics field coupling model was established based on the turbulent bubble flow model by coupling the model of an electric field and fluid heat transfer. The model was used to solve and analyze the bubble rate, temperature, and current density distribution in the machining equilibrium state. Thereafter, the orthogonal experiments were carried out, and the results were obtained. From the results, it was observed the groove contour quality was good, the machining process was stable, and the applied voltage and feed speed had significantly influenced the machining accuracy. Finally, the experiment produced a cross groove with an average entrance width of 2.66 mm and a bottom width of 2.35 mm. -

CGS) Merupakan Beasiswa Full Untuk Meneruskan Jenjang Pendidikan Magister Di Shanghai Normal University Dari Tahun 2016 Sampai Dengan Saat Ini
Departemen Pendidikan dan Pengembangan Organisasi Perhimpunan Pelajar Indonesia (PPI) Tiongkok Beasiswa pertama yang ia peroleh dari Chinese Government Scholarships (CGS) merupakan beasiswa full untuk meneruskan jenjang pendidikan magister di Shanghai Normal University dari tahun 2016 sampai dengan saat ini. Belajar dan tinggal di negeri China merupakan cita-cita kecilnya semenjak usia 6 tahun. Karena ia terinspirasi dari hadits Nabi Muhammad SAW yang berbunyi, “Uthlubul ilma walau bisshin” yang artinya, “Carilah ilmu sampai ke negeri China”. 0 | H a l a m a n Departemen Pendidikan dan Pengembangan Organisasi Perhimpunan Pelajar Indonesia (PPI) Tiongkok 1 | H a l a m a n Departemen Pendidikan dan Pengembangan Organisasi Perhimpunan Pelajar Indonesia (PPI) Tiongkok PPI Tiongkok mempersembahkan PANDUAN KULIAH DI CHINA Edisi 2017 Penulis: Nurul Juliati Putra Wanda Ahmad Fahmi Putri Aris Safitri Tirta anhari Editor: Fadlan Muzakki, Sitti Marwah, Marilyn Janice 2 | H a l a m a n Departemen Pendidikan dan Pengembangan Organisasi Perhimpunan Pelajar Indonesia (PPI) Tiongkok Sambutan Ketua PPI Tiongkok Salam sejahtera untuk kita semua. Ada pepatah yang mengatakan hidup itu harus menjadi garam dan terang bagi dunia. atas dasar itulah PPI Tiongkok ada, yaitu memberikan kontribusi positif bagi Indonesia. PPI Tiongkok sebagai wadah pengembangan minat dan bakat mahasiswa Indonesia memegang peranan penting dalam mempersatukan para pelajar Indonesia di Tiongkok. salah satu misi kami adalah agar para pelajar Indonesia tidak lupa dengan bangsanya sendiri dan suatu saat nanti bisa pulang untuk membangun Indonesia. Oleh karena itu visi Kabinet KB (keluarga berencana) PPI Tiongkok tahun ini adalah memperkuat hubungan internal antar pengurus dan juga meningkatkan kesinergian antara PPIT Cabang. kami yakin dengan hubungan internal yang solid, bahkan hingga mampu menjadi seperti keluarga, ditambah dengan perencanaan yang matang, PPI Tiongkok dapat menghasilkan inisiatif-inisiatif yang dapat berdampak positif bagi mahasiswa-mahasiswa Indonesia yang ada di Tiongkok. -

Fruit Micromorphology in the Umbelliferae of the Russian Far East
Botanica Pacifica. A journal of plant science and conservation. 2018. 7(1): 41–49 DOI: 10.17581/bp.2018.07107 Fruit micromorphology in the Umbelliferae of the Russian Far East Tatiana A. Ostroumova Tatiana A. Ostroumova ABSTRACT e-mail: [email protected] Fruit micromorphology of all 65 native species of the Umbelliferae of the Rus Botanical Garden, Lomonosov Moscow sian Far Easrt was studied. We described cell arangement, cell form, fine relief of state University, Russia cell wall (cuticular foldings), epicuticular wax, stomata, crystals. Scanning electron microscope (SEM) also gave information on the inner structure of plant organs. We discuss the diversity of micromorphological characters and their taxonomic value. Some new diagnostic characters for the genera Kitagawia (areas with rugu Manuscript received: 06.11.2017 late cuticle), Ligusticum and Magadania (large convex exocarp cells) were revealed. Review completed: 14.02.2018 Accepted for publication: 03.04.2018 Keywords: Umbelliferae, taxonomy, identification, SEM Published online: 18.04.2018 РЕЗЮМЕ Остроумова Т.А. Микроморфология плодов зонтичных российско- го Дальнего Востока. Изучена микроморфология плодов всех 65 видов зон тичных Российского Дальнего Востока. Мы описываем расположение клеток, их форму, скульптуру кутикулы, эпикутикулярный воск, устьица, крис таллы. С помощью сканирующего электронного микроскопа возмож но также изучать внутреннюю структуру органов. Обсуждается разнообра зие микроморфологических признаков и их значение для систематики. Для некоторых родов выявлены новые диагностические признаки: участки с морщинистой кутикулой у Kitagawia, крупные выпуклые клетки экзокарпа у Ligusticum и Magadania. Ключевые слова: Umbelliferae, таксономия, определение, сканирующая электрон ная микроскопия INTRODUCTION Fruit micromorphological studies in the Umbelliferae not grai ny. SEM images helped to distinguish between hair showed an importance of these characters for taxonomy types and notice small projections. -

Antioxidant Properties of Angelica Dahurica Extracts Fermented by Probiotics Strains Isolated from Gimchi
Journal of Oil & Applied Science 1 Vol. 35, No. 4. December, 2018. 1276~1284 ISSN 1225-9098 (Print) ISSN 2288-1069 (Online) http://dx.doi.org/10.12925/jkocs.2018.35.4.1276 Antioxidant properties of Angelica dahurica extracts fermented by probiotics strains isolated from gimchi ✝ Joong Gu Ji1*․Sun Kyun Yoo2, 1Department of Oriental Care, Joongbu University 2Department of Food and Biotechnology, Joongbu University (Received November 13, 2018; Revised December 18, 2018; Accepted December 23, 2018) Abstract : probiotics strains promoting the health are a collection of microorganisms that improve or restore microbial populations in the intestines. In this study, Leuconostoc probiotics was isolated from fermented gimchi and identified. Angelica dahurica, containing abundantly antioxidant activity, imperator, is a wildly grown species of angelica native. Before fermentation, total phenolics compound were 48.83 ± 4.9 GAE mg /g in the Angelica dahurica extract. After fermentation total phenolic compounds were 97.7 ± 12.6 GAE mg/g. The total amount of phenol in the fermented product was 30.2% higher than that before fermentation. The total flavonoid content before fermentation was 9.86 ± 4.3 mg / g and the total flavonoid content was 37.17 ± 7.4 mg/g after fermentation, which was 82.3% higher than before fermentation. The DPPH radical scavenging activity, superoxide radical scavenging activity, hydroxy radical scavenging activity and Fe++ chelating antioxidative activity of the Angelica dahurica extract were 41.6 ± 7.1%, 65.7 ± 8.4%, 55.26 ± 9.4% and 17.5 ± 4.6%, respectively. After fermentation, they were 60.3 ± 12.6%, 78.8 ± 8.3%, 56.9 ± 4.9% and 36.6 ± 8.9%, respectively.