Coordination Geometry Primer Waterloo Igem 2020, 21 October
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Inorganic Chemistry
Inorganic Chemistry Chemistry 120 Fall 2005 Prof. Seth Cohen Some Important Features of Metal Ions Electronic configuration • Oxidation State/Charge. •Size. • Coordination number. • Coordination geometry. • “Soft vs. Hard”. • Lability. • Electrochemistry. • Ligand environment. 1 Oxidation State/Charge • The oxidation state describes the charge on the metal center and number of valence electrons • Group - Oxidation Number = d electron count 3 4 5 6 7 8 9 10 11 12 Size • Size of the metal ions follows periodic trends. • Higher positive charge generally means a smaller ion. • Higher on the periodic table means a smaller ion. • Ions of the same charge decrease in radius going across a row, e.g. Ca2+>Mn2+>Zn2+. • Of course, ion size will effect coordination number. 2 Coordination Number • Coordination Number = the number of donor atoms bound to the metal center. • The coordination number may or may not be equal to the number of ligands bound to the metal. • Different metal ions, depending on oxidation state prefer different coordination numbers. • Ligand size and electronic structure can also effect coordination number. Common Coordination Numbers • Low coordination numbers (n =2,3) are fairly rare, in biological systems a few examples are Cu(I) metallochaperones and Hg(II) metalloregulatory proteins. • Four-coordinate is fairly common in complexes of Cu(I/II), Zn(II), Co(II), as well as in biologically less relevant metal ions such as Pd(II) and Pt(II). • Five-coordinate is also fairly common, particularly for Fe(II). • Six-coordinate is the most common and important coordination number for most transition metal ions. • Higher coordination numbers are found in some 2nd and 3rd row transition metals, larger alkali metals, and lanthanides and actinides. -
![Ten-Coordinate Lanthanide [Ln(HL)(L)] Complexes (Ln = Dy, Ho, Er, Tb) with Pentadentate N3O2-Type Schiff-Base Ligands: Synthesis, Structure and Magnetism](https://docslib.b-cdn.net/cover/3551/ten-coordinate-lanthanide-ln-hl-l-complexes-ln-dy-ho-er-tb-with-pentadentate-n3o2-type-schi-base-ligands-synthesis-structure-and-magnetism-673551.webp)
Ten-Coordinate Lanthanide [Ln(HL)(L)] Complexes (Ln = Dy, Ho, Er, Tb) with Pentadentate N3O2-Type Schiff-Base Ligands: Synthesis, Structure and Magnetism
magnetochemistry Article Ten-Coordinate Lanthanide [Ln(HL)(L)] Complexes (Ln = Dy, Ho, Er, Tb) with Pentadentate N3O2-Type Schiff-Base Ligands: Synthesis, Structure and Magnetism Tamara A. Bazhenova 1, Ilya A. Yakushev 1,2,3 , Konstantin A. Lyssenko 4 , Olga V. Maximova 1,5,6, Vladimir S. Mironov 1,7,* , Yuriy V. Manakin 1 , Alexey B. Kornev 1, Alexander N. Vasiliev 5,8 and Eduard B. Yagubskii 1,* 1 Institute of Problems of Chemical Physics, 142432 Chernogolovka, Russia; [email protected] (T.A.B.); [email protected] (I.A.Y.); [email protected] (O.V.M.); [email protected] (Y.V.M.); [email protected] (A.B.K.) 2 Kurnakov Institute of General and Inorganic Chemistry, 119991 Moscow, Russia 3 National Research Center “Kurchatov Institute”, 123182 Moscow, Russia 4 Department of Chemistry, Lomonosov Moscow State University, 119991 Moscow, Russia; [email protected] 5 Faculty of Physics, Lomonosov Moscow State University, 119991 Moscow, Russia; [email protected] 6 National University “MISiS”, 119049 Moscow, Russia 7 Shubnikov Institute of Crystallography of Federal Scientific Research Centre “Crystallography and Photonics” RAS, 119333 Moscow, Russia 8 National Research South Ural State University, 454080 Chelyabinsk, Russia * Correspondence: [email protected] (V.S.M.); [email protected] (E.B.Y.) Received: 26 October 2020; Accepted: 9 November 2020; Published: 13 November 2020 Abstract: A series of five neutral mononuclear lanthanide complexes [Ln(HL)(L)] (Ln = Dy3+, 3+ 3+ 3+ H Ho Er and Tb ) with rigid pentadentate N3O2-type Schiff base ligands, H2L (1-Dy, OCH3 3-Ho, 4-Er and 6-Tb complexes) or H2L ,(2-Dy complex) has been synthesized by reaction H of two equivalents of 1,10-(pyridine-2,6-diyl)bis(ethan-1-yl-1-ylidene))dibenzohydrazine (H2L , [H2DAPBH]) or 1,10-(pyridine-2,6-diyl)bis(ethan-1-yl-1-ylidene))di-4-methoxybenzohydrazine OCH3 (H2L , [H2DAPMBH]) with common lanthanide salts. -

Bicapped Tetrahedral, Trigonal Prismatic, and Octahedral Alternatives in Main and Transition Group Six-Coordination
2484 Bicapped Tetrahedral, Trigonal Prismatic, and Octahedral Alternatives in Main and Transition Group Six-Coordination Roald Hoffmann,*laJames M. Howell,lb and Angelo R. RossilC Contribution from the Departments of Chemistry, Cornell University, Ithaca, New York 14853, Brooklyn College, City University of New York. Brooklyn, New York, 1121 0, and The University of Connecticut, Storrs, Connecticut 06268. Received June 30, 1975 Abstract: We examine theoretically the bicapped tetrahedron and trigonal prism as alternatives to the common octahedron for six-coordinate main and transition group compounds. The pronounced preference of six-coordinate sulfur for an octahe- dral geometry is traced by a molecular orbital analysis to a pair of nonbonding levels, whose higher energy in the nonoctahe- dral geometries is due to the molecular orbital equivalent of a ligand-ligand repulsion. For transition element complexes we draw a correlation diagram for the trigonal twist interrelating octahedral and trigonal prismatic extremes. A possible prefer- ence for the trigonal prism in systems with few d electrons is discussed, as well as a strategy for lowering the energy of the trigonal prism by symmetry-conditioned 7r bonding. The bicapped tetrahedron geometry for transition metal complexes can be stabilized by a substitution pattern ML4D2 where D is a good u donor and M is a d6 metal center. In main and transition group six-coordinate compounds logical, or group theoretical analysis of rearrangement the octahedral geometry predominates. There is, however, a modes or basic permutational sets. However, the solution of growing class of trigonal prismatic complexes and struc- the mathematical problem leaves the question of the physi- tures intermediate between the octahedron and trigonal cal mechanism of a rearrangement still unsolved. -

The Transition Metals
The Transition Metals • d electrons in group 3 are readily removed via ionization . • d electrons in group 11 are stable and generally form part of the core electron configuration. Transition Metal Valence Orbitals • nd orbitals • (n + 1)s and (n + 1)p orbitals • 2 2 2 dx -dy and dz (e g) lobes located on the axes • dxy, dxz, dyz lobes (t 2g) located between axes • orbitals oriented orthogonal wrt each other creating unique possibilities for ligand overlap. • Total of 9 valence orbitals available for bonding (2 x 9 = 18 valence electrons!) • For an σ bonding only Oh complex, 6 σ bonds are formed and the remaining d orbitals are non-bonding . • It's these non-bonding d orbitals that give TM complexes many of their unique properties • for free (gas phase) transition metals: (n+1)s is below (n)d in energy (recall: n = principal quantum #). • for complexed transition metals: the (n)d levels are below the (n+1)s and thus get filled first. (note that group # = d electron count) • for oxidized metals, subtract the oxidation state from the group #. Geometry of Transition Metals Coordination Geometry – arrangement of ligands around metal centre Valence Shell Electron Pair Repulsion (VSEPR) theory is generally not applicable to transition metals complexes (ligands still repel each other as in VSEPR theory) For example, a different geometry would be expected for metals of different d electron count 3+ 2 [V(OH 2)6] d 3+ 4 [Mn(OH 2)6] d all octahedral geometry ! 3+ 6 [Co(OH 2)6] d Coordination geometry is , in most cases , independent of ground state electronic configuration Steric: M-L bonds are arranged to have the maximum possible separation around the M. -

IR-9 Coordination Compounds (Draft March 2004)
1 IR-9 Coordination Compounds (Draft March 2004) CONTENTS IR-9.1 Introduction IR-9.1.1 General IR-9.1.2 Definitions IR-9.1.2.1 Background IR-9.1.2.2 Coordination compounds and the coordination entity IR-9.1.2.3 Central atom IR-9.1.2.4 Ligands IR-9.1.2.5 Coordination polyhedron IR-9.1.2.6 Coordination number IR-9.1.2.7 Chelation IR-9.1.2.8 Oxidation state IR-9.1.2.9 Coordination nomenclature: an additive nomenclature IR-9.1.2.10 Bridging ligands IR-9.1.2.11 Metal-metal bonds IR-9.2 Describing the constitution of coordination compounds IR-9.2.1 General IR-9.2.2 Naming coordination compounds IR-9.2.2.1 Sequences of ligands and central atoms within names IR-9.2.2.2 Number of ligands in a coordination entity IR-9.2.2.3 Representing ligands in names IR-9.2.2.4 Charge numbers, oxidation numbers, and ionic proportions IR-9.2.3 Formulae of coordination compounds IR-9.2.3.1 Sequence of symbols within the coordination formula IR-9.2.3.2 Use of enclosing marks IR-9.2.3.3 Ionic charges and oxidation numbers IR-9.2.3.4 Use of abbreviations IR-9.2.4 Specifying donor atoms IR-9.2.4.1 General IUPAC ProvisionalIR-9.2.4.2 The kappa convention Recommendations IR-9.2.4.3 Comparison of the eta and kappa nomenclatures Page 1 of 69 DRAFT 2 April 2004 2 IR-9.2.4.4 Donor atom symbol to indicate points of ligation IR-9.2.5 Polynuclear complexes IR-9.2.5.1 General IR-9.2.5.2 Bridging ligands IR-9.2.5.3 Metal-metal bonding IR-9.2.5.4 Symmetrical dinuclear entities IR-9.2.5.5 Unsymmetrical dinuclear entities IR-9.2.5.6 Trinuclear and larger structures IR-9.2.5.7 Polynuclear -

Eight-Coordination Inorganic Chemistry, Vol
Eight-Coordination Inorganic Chemistry, Vol. 17, No. 9, 1978 2553 Contribution from the Department of Chemistry, Cornell University, Ithaca, New York 14853 Eight-Coordination JEREMY K. BURDETT,’ ROALD HOFFMANN,* and ROBERT C. FAY Received April 14, 1978 A systematic molecular orbital analysis of eight-coordinate molecules is presented. The emphasis lies in appreciating the basic electronic structure, u and ?r substituent site preferences, and relative bond lengths within a particular geometry for the following structures: dodecahedron (DD), square antiprism (SAP),C, bicapped trigonal prism (BTP), cube (C), hexagonal bipyramid (HB), square prism (SP), bicapped trigonal antiprism (BTAP), and Dgh bicapped trigonal prism (ETP). With respect to u or electronegativity effects the better u donor should lie in the A sites of the DD and the capping sites of the BTP, although the preferences are not very strong when viewed from the basis of ligand charges. For d2P acceptors and do ?r donors, a site preferences are Ig> LA> IIA,B (> means better than) for the DD (this is Orgel’s rule), 11 > I for the SAP, (mil- bll) > bl > (cil N cI N m,) for the BTP (b, c, and m refer to ligands which are basal, are capping, or belong to the trigonal faces and lie on a vertical mirror plane) and eqli- ax > eq, for the HB. The reverse order holds for dZdonors. The observed site preferences in the DD are probably controlled by a mixture of steric and electronic (u, a) effects. An interesting crossover from r(M-A)/r(M-B) > 1 to r(M-A)/r(M-B) < 1 is found as a function of geometry for the DD structure which is well matched by experimental observations. -

Structures of Metal–Organic Frameworks with Rod Secondary Building Units
Review pubs.acs.org/CR Structures of Metal−Organic Frameworks with Rod Secondary Building Units † ‡ § ∥ ∥ ⊥ † ‡ ▽ Alexander Schoedel, , , Mian Li, Dan Li, , Michael O’Keeffe,*,@ and Omar M. Yaghi , , † Department of Chemistry, University of California, Berkeley, California 94720, United States ‡ Materials Sciences Division, Lawrence Berkeley National Laboratory, Kavli Energy Nanoscience Institute, Berkeley, California 94720, United States § Department of Chemistry, Florida Institute of Technology, 150 West University Boulevard, Melbourne, Florida 32901, United States ∥ Department of Chemistry, Shantou University, Guangdong 515063, P. R. China ⊥ College of Chemistry and Materials Science, Jinan University, Guangzhou 510632, P. R. China @School of Molecular Sciences, Arizona State University, Tempe, Arizona 85287, United States ▽ King Abdulaziz City for Science and Technology, P.O Box 6086, Riyadh 11442, Saudi Arabia *S Supporting Information ABSTRACT: Rod MOFs are metal−organic frameworks in which the metal-containing secondary building units consist of infinite rods of linked metal-centered polyhedra. For such materials, we identify the points of extension, often atoms, which define the interface between the organic and inorganic components of the structure. The pattern of points of extension defines a shape such as a helix, ladder, helical ribbon, or cylinder tiling. The linkage of these shapes into a three-dimensional framework in turn defines a net characteristic of the original structure. Some scores of rod MOF structures are illustrated and deconstructed into their underlying nets in this way. Crystallographic data for all nets in their maximum symmetry embeddings are provided. CONTENTS 5. MOFs with SBUs of Edge- or Face-Shared Octahedra W 1. Introduction B − 5.1. MOFs with SBUs of Edge- or Face-Shared 1.1. -

The Rise of Metal–Organic Polyhedra
Chem Soc Rev View Article Online REVIEW ARTICLE View Journal | View Issue The rise of metal–organic polyhedra Cite this: Chem. Soc. Rev.,2021, Soochan Lee, Hyein Jeong, Dongsik Nam, Myoung Soo Lah * and 50,528 Wonyoung Choe * Metal–organic polyhedra are a member of metal–organic materials, and are together with metal– organic frameworks utilized as emerging porous platforms for numerous applications in energy- and Received 2nd July 2020 bio-related sciences. However, metal–organic polyhedra have been significantly underexplored, unlike DOI: 10.1039/d0cs00443j their metal–organic framework counterparts. In this review, we will cover the topologies and the classifi- cation of metal–organic polyhedra and share several suggestions, which might be useful to synthetic rsc.li/chem-soc-rev chemists regarding the future directions in this rapid-growing field. 1. Introduction (octahedron), fire (tetrahedron), water (icosahedron), and ether (dodecahedron) in the dialogue Timaeus.4 Kepler modeled Polyhedra were discovered in the early stages of civilization in the solar system as nested polyhedra in his book Mysterium the east and the west and are found in art, science, architecture, Cosmographicum, reflecting his theological ideas.5 It is striking and humanities to describe not only tangible objects but also to note that a report in 2003 has suggested a dodecahedron as metaphysical concepts, thus becoming a form of language. the shape of the universe.6 Scientists have discovered polyhedra Some of the interesting examples are as follows. Polyhedron- -
Coordination Chemistry of Vanadium Aquo Complex Ions in Oxidation States +II, +III, +IV, and +V: a Hybrid-Functional DFT Study
769 International Journal of Progressive Sciences and Technologies (IJPSAT) ISSN: 2509-0119. © 2020 International Journals of Sciences and High Technologies http://ijpsat.ijsht-journals.org Vol. 24 No. 1 December 2020, pp. 645-661 Coordination Chemistry of Vanadium Aquo Complex Ions in Oxidation States +II, +III, +IV, and +V: A Hybrid-Functional DFT Study Anant Babu Marahatta1, 2 1Department of Chemistry, Amrit Science Campus, Tribhuvan University, Kathmandu, Nepal 2Chemistry Subject Committee, Kathford International College of Engineering and Management (Affiliated to Tribhuvan University), Kathmandu, Nepal 2+ 3+ 2+ + n+ Abstract – The vanadium aquo complexes: [V(H2O)6] , [V(H2O)6] , [VO(H2O)5] , and [VO2(H2O)3] . H2O containing V : n = +II, +III, +IV, and +V ion respectively with only H2O as ligands are the most prevailing ionic species in their aqueous type medicinal and biological fluid matrices, and all-vanadium redox flow battery (VRFB) systems. Since, they tend to display particular configurations with distinctive electronic stabilities, understanding how each adjacent vanadium ion stabilizes itself with specific hydration number and acquires unique equilibrium structure is very indispensable. With this as a major objective, the coordination chemistry of all these four hydrated vanadium complexes are studied here thoroughly by applying a hybrid-functional DFT method. It is found that all the theoretically derived bond 2+ lengths (Th.) of each optimized complex ion agree reasonably with the experimental values (Exp.): (a) [V(H2O)6] : V−OH2 Th. 2.0 Å, Exp. 3+ 2+ 2.1 Å; (b) [V(H2O)6] : V−OH2 Th. 1.98 Å, Exp. 1.99 Å; (c) [VO(H2O)5] : equatorial V−OH2 Th. -
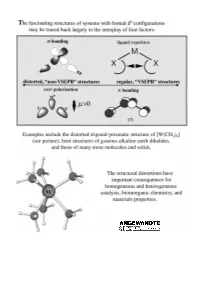
"Non-VSEPR" Structures and Bonding in D0 Systems
REVIEWS ªNon-VSEPRº Structures and Bonding in d0 Systems Martin Kaupp* Dedicated to the memory of Marco Häser (1961 ± 1997) Under certain circumstances, metal denum or tungsten enzymes), or mate- d) p bonding. Suggestions are made complexes with a formal d0 electronic rials science (e.g. ferroelectric as to which of the factors are the configuration may exhibit structures perovskites or zirconia). Moreover, dominant ones in certain situations. that violate the traditional structure their electronic structure without for- In heteroleptic complexes, the compe- models, such as the VSEPR concept or mally nonbonding d orbitals makes tition of s and p bonding of the various simple ionic pictures. Some examples them unique starting points for a gen- ligands controls the structures in a of such behavior, such as the bent gas- eral understanding of structure, bond- complicated fashion. Some guidelines phase structures of some alkaline earth ing, and reactivity of transition metal are provided that should help to better dihalides, or the trigonal prismatic compounds. Here we attempt to pro- understand the interrelations. Bents coordination of some early transition vide a comprehensive view, both of the rule is of only very limited use in these metal chalcogenides or pnictides, have types of deviations of d0 and related types of systems, because of the para- been known for a long time. However, complexes from regular coordination mount influence of p bonding. Finally, the number of molecular examples for arrangements, and of the theoretical computed and measured structures of ªnon-VSEPRº structures has in- framework that allows their rationali- multinuclear complexes are discussed, creased dramatically during the past zation. -

Chapter 4. Systematic Studies of Geometry Of
Durham E-Theses Systematic structural studies in metal complex chemistry Yao, Jing Wen How to cite: Yao, Jing Wen (1998) Systematic structural studies in metal complex chemistry, Durham theses, Durham University. Available at Durham E-Theses Online: http://etheses.dur.ac.uk/5055/ Use policy The full-text may be used and/or reproduced, and given to third parties in any format or medium, without prior permission or charge, for personal research or study, educational, or not-for-prot purposes provided that: • a full bibliographic reference is made to the original source • a link is made to the metadata record in Durham E-Theses • the full-text is not changed in any way The full-text must not be sold in any format or medium without the formal permission of the copyright holders. Please consult the full Durham E-Theses policy for further details. Academic Support Oce, Durham University, University Oce, Old Elvet, Durham DH1 3HP e-mail: [email protected] Tel: +44 0191 334 6107 http://etheses.dur.ac.uk Systematic Structural Studies Metal Complex Chemistry by Jing Wen Yao The copyright of this thesis rests with the author. No quotation from it should be published without the written consent of the author and information derived from it should be acknowledged. A thesis submitted for the degree of Doctor of Philosophy Department of Chemistry- University of Durham November, 1998 16 Contents List of Figures iv List of Tables viii Acknowledgements x Declaration xi Abstract xii Chapter 1. Introduction 1 Chapter 2. Structural Systematics: the -

An Introduction to Transition Metal Chemistry
An Introduction to Transition Metal Chemistry A transition metal is an element that has a partly d or f subshell in any of its common oxidation states. The transition elements are taken to be those of Groups 3-12, plus the lanthanides and actinides. Properties Common to the Transition Elements 1. The free elements conform to the metallic bonding model. Thus, their lattices are typically either close-packed or body centered cubic. They generally have high thermal and electrical conductivities, and they are malleable and ductile. 2. Nearly all have more that one positive oxidation state. 3. Nearly all have one or more unpaired electrons in their atomic ground states, and form paramagnetic compounds and ions. Because of this, studies of magnetic properties using experimental tools such as NMR, ESR, and magnetic susceptibility are often employed in this are of chemistry. 4. Low-energy electron transitions are observed for the free elements and. their compounds and complexes. The transitions may fall into the infrared, visible, or ultraviolet region; for the visible cases, of course, colored species are the results. 5. The cations of these elements (and often the neutral atoms as well) behave as Lewis acids, and there is a strong tendency to form complexes. Complexes having 2-6 bases (ligands) are common, and species with as many as 14 ligands about a single metal are known. Oxidation State Tendencies and Their Causes A key to understanding the chemistry of the transition elements is knowledge of their oxidation state preferences. These preferences can be rationalized through analysis of such factors as ionization energy (IE) and bond energy.