Identifying Endogenous Binding Partners for Btf and Trap150
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Urabe VK, Et Al. Influences on U2 Snrna Structure U2
bioRxiv preprint doi: https://doi.org/10.1101/2021.07.05.451154; this version posted July 6, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Urabe VK, et al. Influences on U2 snRNA structure U2 snRNA structure is influenced by SF3A and SF3B proteins but not by SF3B inhibitors Veronica K. Urabe1, Meredith Stevers1, Arun K. Ghosh3 and Melissa S. Jurica1,2 * 1Department of Molecular Cell and Developmental Biology and 2Center for Molecular Biology of RNA, University of California, Santa Cruz, California, United States of America 3Department of Chemistry and Department of Medicinal Chemistry, Purdue University, West Lafayette, Indiana United States of America *Corresponding author E-mail: [email protected] (MSJ) bioRxiv preprint doi: https://doi.org/10.1101/2021.07.05.451154; this version posted July 6, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Urabe VK, et al. Influences on U2 snRNA structure Abstract U2 snRNP is an essential component of the spliceosome. It is responsible for branch point recognition in the spliceosome A-complex via base-pairing of U2 snRNA with an intron to form the branch helix. Small molecule inhibitors target the SF3B component of the U2 snRNP and interfere with A-complex formation during spliceosome assembly. -

SF3B2 Monoclonal Antibody (M01J), Clone 5D2
SF3B2 monoclonal antibody (M01J), clone 5D2 Catalog # : H00010992-M01J 規格 : [ 100 ug ] List All Specification Application Image Product Mouse monoclonal antibody raised against a partial recombinant Western Blot (Cell lysate) Description: SF3B2. Immunogen: SF3B2 (NP_006833, 592 a.a. ~ 645 a.a) partial recombinant protein with GST tag. MW of the GST tag alone is 26 KDa. Sequence: YEGKEFETRLKEKKPGDLSDELRISLGMPVGPNAHKVPPPWLIAMQRYG PPPSY enlarge Western Blot (Cell lysate) Host: Mouse Reactivity: Human, Mouse, Rat Preparation Cell Culture Production Method: (CX Grade Antibody List) enlarge Isotype: IgG2a Kappa Western Blot (Recombinant protein) Quality Control Antibody Reactive Against Recombinant Protein. Immunofluorescence Testing: enlarge Immunohistochemistry (Formalin/PFA-fixed paraffin- embedded sections) Western Blot detection against Immunogen (31.68 KDa) . Storage Buffer: In 1x PBS, pH 7.4 Storage Store at -20°C or lower. Aliquot to avoid repeated freezing and thawing. enlarge Instruction: Sandwich ELISA (Recombinant protein) MSDS: Download Interspecies Mouse (98); Rat (98) Antigen Sequence: Datasheet: Download enlarge ELISA Applications Western Blot (Cell lysate) Page 1 of 3 2021/6/15 SF3B2 monoclonal antibody (M01J), clone 5D2. Western Blot analysis of SF3B2 expression in Hela S3 NE. Protocol Download Western Blot (Cell lysate) SF3B2 monoclonal antibody (M01J), clone 5D2. Western Blot analysis of SF3B2 expression in Jurkat. Protocol Download Western Blot (Recombinant protein) Protocol Download Immunofluorescence enlarge this image Immunofluorescence of monoclonal antibody to SF3B2 on HeLa cell . [antibody concentration 10 ug/ml] Immunohistochemistry (Formalin/PFA-fixed paraffin-embedded sections) enlarge this image Page 2 of 3 2021/6/15 Immunoperoxidase of monoclonal antibody to SF3B2 on formalin-fixed paraffin-embedded human kidney. [antibody concentration 6 ug/ml] Protocol Download Sandwich ELISA (Recombinant protein) Detection limit for recombinant GST tagged SF3B2 is 0.1 ng/ml as a capture antibody. -

SF3B2-Mediated RNA Splicing Drives Human Prostate Cancer Progression
Published OnlineFirst August 20, 2019; DOI: 10.1158/0008-5472.CAN-18-3965 Cancer Molecular Cell Biology Research SF3B2-Mediated RNA Splicing Drives Human Prostate Cancer Progression Norihiko Kawamura1,2, Keisuke Nimura1, Kotaro Saga1, Airi Ishibashi1, Koji Kitamura1,3, Hiromichi Nagano1, Yusuke Yoshikawa4, Kyoso Ishida1,5, Norio Nonomura2, Mitsuhiro Arisawa4, Jun Luo6, and Yasufumi Kaneda1 Abstract Androgen receptor splice variant-7 (AR-V7) is a General RNA splicing SF3B2 complex-mediated alternative RNA splicing constitutively active AR variant implicated in U2 castration-resistant prostate cancers. Here, we show U2 snRNA that the RNA splicing factor SF3B2, identified by 3’ 3’ in silico and CRISPR/Cas9 analyses, is a critical 5’ 3’ splice site 5’ SF3B7 AR-V7 5’ A U2AF2 AGA Exon ? determinant of expression and is correlated SF3B6(p14) SF3B4 SF3B1 SF3B4 SF3B1 with aggressive cancer phenotypes. Transcriptome SF3B5 SF3B2 SF3B3 SF3B2 SF3B3 and PAR-CLIP analyses revealed that SF3B2 con- SF3A3 SF3B2 complex SF3A3 SF3A1 SF3A1 SF3b complex trols the splicing of target genes, including AR, to AR pre-mRNA drive aggressive phenotypes. SF3B2-mediated CE3 aggressive phenotypes in vivo were reversed by AR-V7 mRNA AR mRNA AR-V7 knockout. Pladienolide B, an inhibitor of CE3 a splicing modulator of the SF3b complex, sup- Drive malignancy pressed the growth of tumors addicted to high While the SF3b complex is critical for general RNA splicing, SF3B2 promotes inclusion of the target exon through recognizing a specific RNA motif. SF3B2 expression. These findings support the idea © 2019 American Association for Cancer Research that alteration of the splicing pattern by high SF3B2 expression is one mechanism underlying prostate cancer progression and therapeutic resistance. -

Essential Genes and Their Role in Autism Spectrum Disorder
University of Pennsylvania ScholarlyCommons Publicly Accessible Penn Dissertations 2017 Essential Genes And Their Role In Autism Spectrum Disorder Xiao Ji University of Pennsylvania, [email protected] Follow this and additional works at: https://repository.upenn.edu/edissertations Part of the Bioinformatics Commons, and the Genetics Commons Recommended Citation Ji, Xiao, "Essential Genes And Their Role In Autism Spectrum Disorder" (2017). Publicly Accessible Penn Dissertations. 2369. https://repository.upenn.edu/edissertations/2369 This paper is posted at ScholarlyCommons. https://repository.upenn.edu/edissertations/2369 For more information, please contact [email protected]. Essential Genes And Their Role In Autism Spectrum Disorder Abstract Essential genes (EGs) play central roles in fundamental cellular processes and are required for the survival of an organism. EGs are enriched for human disease genes and are under strong purifying selection. This intolerance to deleterious mutations, commonly observed haploinsufficiency and the importance of EGs in pre- and postnatal development suggests a possible cumulative effect of deleterious variants in EGs on complex neurodevelopmental disorders. Autism spectrum disorder (ASD) is a heterogeneous, highly heritable neurodevelopmental syndrome characterized by impaired social interaction, communication and repetitive behavior. More and more genetic evidence points to a polygenic model of ASD and it is estimated that hundreds of genes contribute to ASD. The central question addressed in this dissertation is whether genes with a strong effect on survival and fitness (i.e. EGs) play a specific oler in ASD risk. I compiled a comprehensive catalog of 3,915 mammalian EGs by combining human orthologs of lethal genes in knockout mice and genes responsible for cell-based essentiality. -

Myopia in African Americans Is Significantly Linked to Chromosome 7P15.2-14.2
Genetics Myopia in African Americans Is Significantly Linked to Chromosome 7p15.2-14.2 Claire L. Simpson,1,2,* Anthony M. Musolf,2,* Roberto Y. Cordero,1 Jennifer B. Cordero,1 Laura Portas,2 Federico Murgia,2 Deyana D. Lewis,2 Candace D. Middlebrooks,2 Elise B. Ciner,3 Joan E. Bailey-Wilson,1,† and Dwight Stambolian4,† 1Department of Genetics, Genomics and Informatics and Department of Ophthalmology, University of Tennessee Health Science Center, Memphis, Tennessee, United States 2Computational and Statistical Genomics Branch, National Human Genome Research Institute, National Institutes of Health, Baltimore, Maryland, United States 3The Pennsylvania College of Optometry at Salus University, Elkins Park, Pennsylvania, United States 4Department of Ophthalmology, University of Pennsylvania, Philadelphia, Pennsylvania, United States Correspondence: Joan E. PURPOSE. The purpose of this study was to perform genetic linkage analysis and associ- Bailey-Wilson, NIH/NHGRI, 333 ation analysis on exome genotyping from highly aggregated African American families Cassell Drive, Suite 1200, Baltimore, with nonpathogenic myopia. African Americans are a particularly understudied popula- MD 21131, USA; tion with respect to myopia. [email protected]. METHODS. One hundred six African American families from the Philadelphia area with a CLS and AMM contributed equally to family history of myopia were genotyped using an Illumina ExomePlus array and merged this work and should be considered co-first authors. with previous microsatellite data. Myopia was initially measured in mean spherical equiv- JEB-W and DS contributed equally alent (MSE) and converted to a binary phenotype where individuals were identified as to this work and should be affected, unaffected, or unknown. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

Original Article a Database and Functional Annotation of NF-Κb Target Genes
Int J Clin Exp Med 2016;9(5):7986-7995 www.ijcem.com /ISSN:1940-5901/IJCEM0019172 Original Article A database and functional annotation of NF-κB target genes Yang Yang, Jian Wu, Jinke Wang The State Key Laboratory of Bioelectronics, Southeast University, Nanjing 210096, People’s Republic of China Received November 4, 2015; Accepted February 10, 2016; Epub May 15, 2016; Published May 30, 2016 Abstract: Backgrounds: The previous studies show that the transcription factor NF-κB always be induced by many inducers, and can regulate the expressions of many genes. The aim of the present study is to explore the database and functional annotation of NF-κB target genes. Methods: In this study, we manually collected the most complete listing of all NF-κB target genes identified to date, including the NF-κB microRNA target genes and built the database of NF-κB target genes with the detailed information of each target gene and annotated it by DAVID tools. Results: The NF-κB target genes database was established (http://tfdb.seu.edu.cn/nfkb/). The collected data confirmed that NF-κB maintains multitudinous biological functions and possesses the considerable complexity and diversity in regulation the expression of corresponding target genes set. The data showed that the NF-κB was a central regula- tor of the stress response, immune response and cellular metabolic processes. NF-κB involved in bone disease, immunological disease and cardiovascular disease, various cancers and nervous disease. NF-κB can modulate the expression activity of other transcriptional factors. Inhibition of IKK and IκBα phosphorylation, the decrease of nuclear translocation of p65 and the reduction of intracellular glutathione level determined the up-regulation or down-regulation of expression of NF-κB target genes. -

Structural Insights Into How Prp5 Proofreads the Pre-Mrna Branch Site
Article Structural insights into how Prp5 proofreads the pre-mRNA branch site https://doi.org/10.1038/s41586-021-03789-5 Zhenwei Zhang1, Norbert Rigo2, Olexandr Dybkov2, Jean-Baptiste Fourmann2, Cindy L. Will2, Vinay Kumar2, Henning Urlaub3,4, Holger Stark1 ✉ & Reinhard Lührmann2 ✉ Received: 10 December 2020 Accepted: 30 June 2021 During the splicing of introns from precursor messenger RNAs (pre-mRNAs), the U2 Published online: 4 August 2021 small nuclear ribonucleoprotein (snRNP) must undergo stable integration into the Open access spliceosomal A complex—a poorly understood, multistep process that is facilitated by Check for updates the DEAD-box helicase Prp5 (refs. 1–4). During this process, the U2 small nuclear RNA (snRNA) forms an RNA duplex with the pre-mRNA branch site (the U2–BS helix), which is proofread by Prp5 at this stage through an unclear mechanism5. Here, by deleting the branch-site adenosine (BS-A) or mutating the branch-site sequence of an actin pre-mRNA, we stall the assembly of spliceosomes in extracts from the yeast Saccharomyces cerevisiae directly before the A complex is formed. We then determine the three-dimensional structure of this newly identifed assembly intermediate by cryo-electron microscopy. Our structure indicates that the U2–BS helix has formed in this pre-A complex, but is not yet clamped by the HEAT domain of the Hsh155 protein (Hsh155HEAT), which exhibits an open conformation. The structure further reveals a large-scale remodelling/repositioning of the U1 and U2 snRNPs during the formation of the A complex that is required to allow subsequent binding of the U4/U6.U5 tri-snRNP, but that this repositioning is blocked in the pre-A complex by the presence of Prp5. -
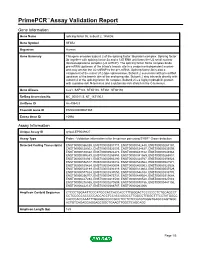
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name splicing factor 3b, subunit 2, 145kDa Gene Symbol SF3B2 Organism Human Gene Summary This gene encodes subunit 2 of the splicing factor 3b protein complex. Splicing factor 3b together with splicing factor 3a and a 12S RNA unit forms the U2 small nuclear ribonucleoproteins complex (U2 snRNP). The splicing factor 3b/3a complex binds pre-mRNA upstream of the intron's branch site in a sequence-independent manner and may anchor the U2 snRNP to the pre-mRNA. Splicing factor 3b is also a component of the minor U12-type spliceosome. Subunit 2 associates with pre-mRNA upstream of the branch site at the anchoring site. Subunit 2 also interacts directly with subunit 4 of the splicing factor 3b complex. Subunit 2 is a highly hydrophilic protein with a proline-rich N-terminus and a glutamate-rich stretch in the C-terminus. Gene Aliases Cus1, SAP145, SF3B145, SF3b1, SF3b150 RefSeq Accession No. NC_000011.9, NT_167190.1 UniGene ID Hs.406423 Ensembl Gene ID ENSG00000087365 Entrez Gene ID 10992 Assay Information Unique Assay ID qHsaCEP0049827 Assay Type Probe - Validation information is for the primer pair using SYBR® Green detection Detected Coding Transcript(s) ENST00000566339, ENST00000301717, ENST00000542440, ENST00000267197, ENST00000528302, ENST00000322535, ENST00000524627, ENST00000533595, ENST00000530322, ENST00000524475, ENST00000483722, ENST00000545968, ENST00000399249, ENST00000256993, ENST00000381569, ENST00000265801, ENST00000538941, ENST00000563290, ENST00000324767, ENST00000356924, -

Combinatorial Strategies Using CRISPR/Cas9 for Gene Mutagenesis in Adult Mice
Combinatorial strategies using CRISPR/Cas9 for gene mutagenesis in adult mice Avery C. Hunker A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy University of Washington 2019 Reading Committee: Larry S. Zweifel, Chair Sheri J. Mizumori G. Stanley McKnight Program Authorized to Offer Degree: Pharmacology 2 © Copyright 2019 Avery C. Hunker 3 University of Washington ABSTRACT Combinatorial strategies using CRISPR/Cas9 for gene mutagenesis in adult mice Avery C. Hunker Chair of the Supervisory Committee: Larry Zweifel Department of Pharmacology A major challenge to understanding how genes modulate complex behaviors is the inability to restrict genetic manipulations to defined cell populations or circuits. To circumvent this, we created a simple strategy for limiting gene knockout to specific cell populations using a viral-mediated, conditional CRISPR/SaCas9 system in combination with intersectional genetic strategies. A small single guide RNA (sgRNA) directs Staphylococcus aureus CRISPR-associated protein (SaCas9) to unique sites on DNA in a Cre-dependent manner resulting in double strand breaks and gene mutagenesis in vivo. To validate this technique we targeted nine different genes of diverse function in distinct cell types in mice and performed an array of analyses to confirm gene mutagenesis and subsequent protein loss, including IHC, cell-type specific DNA sequencing, electrophysiology, Western blots, and behavior. We show that these vectors are as efficient as conventional conditional gene knockout and provide a viable alternative to complex genetic crosses. This strategy provides additional benefits of 4 targeting gene mutagenesis to cell types previously difficult to isolate, and the ability to target genes in specific neural projections for gene inactivation. -

The RNA Splicing Response to DNA Damage
Biomolecules 2015, 5, 2935-2977; doi:10.3390/biom5042935 OPEN ACCESS biomolecules ISSN 2218-273X www.mdpi.com/journal/biomolecules/ Review The RNA Splicing Response to DNA Damage Lulzim Shkreta and Benoit Chabot * Département de Microbiologie et d’Infectiologie, Faculté de Médecine et des Sciences de la Santé, Université de Sherbrooke, Sherbrooke, QC J1E 4K8, Canada; E-Mail: [email protected] * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +1-819-821-8000 (ext. 75321); Fax: +1-819-820-6831. Academic Editors: Wolf-Dietrich Heyer, Thomas Helleday and Fumio Hanaoka Received: 12 August 2015 / Accepted: 16 October 2015 / Published: 29 October 2015 Abstract: The number of factors known to participate in the DNA damage response (DDR) has expanded considerably in recent years to include splicing and alternative splicing factors. While the binding of splicing proteins and ribonucleoprotein complexes to nascent transcripts prevents genomic instability by deterring the formation of RNA/DNA duplexes, splicing factors are also recruited to, or removed from, sites of DNA damage. The first steps of the DDR promote the post-translational modification of splicing factors to affect their localization and activity, while more downstream DDR events alter their expression. Although descriptions of molecular mechanisms remain limited, an emerging trend is that DNA damage disrupts the coupling of constitutive and alternative splicing with the transcription of genes involved in DNA repair, cell-cycle control and apoptosis. A better understanding of how changes in splice site selection are integrated into the DDR may provide new avenues to combat cancer and delay aging. -

Roles of Splicing Factors in Hormone-Related Cancer Progression
International Journal of Molecular Sciences Review Roles of Splicing Factors in Hormone-Related Cancer Progression Toshihiko Takeiwa 1, Yuichi Mitobe 1, Kazuhiro Ikeda 1, Kuniko Horie-Inoue 1 and Satoshi Inoue 1,2,* 1 Division of Gene Regulation and Signal Transduction, Research Center for Genomic Medicine, Saitama Medical University, Hidaka, Saitama 350-1241, Japan; [email protected] (T.T.); [email protected] (Y.M.); [email protected] (K.I.); [email protected] (K.H.-I.) 2 Department of Systems Aging Science and Medicine, Tokyo Metropolitan Institute of Gerontology, Itabashi-ku, Tokyo 173-0015, Japan * Correspondence: [email protected]; Tel.: +81-3-3964-3241 Received: 8 February 2020; Accepted: 20 February 2020; Published: 25 February 2020 Abstract: Splicing of mRNA precursor (pre-mRNA) is a mechanism to generate multiple mRNA isoforms from a single pre-mRNA, and it plays an essential role in a variety of biological phenomena and diseases such as cancers. Previous studies have demonstrated that cancer-specific splicing events are involved in various aspects of cancers such as proliferation, migration and response to hormones, suggesting that splicing-targeting therapy can be promising as a new strategy for cancer treatment. In this review, we focus on the splicing regulation by RNA-binding proteins including Drosophila behavior/human splicing (DBHS) family proteins, serine/arginine-rich (SR) proteins and heterogeneous nuclear ribonucleoproteins (hnRNPs) in hormone-related cancers, such as breast and prostate cancers. Keywords: DBHS family proteins; SR proteins; hnRNPs; breast cancer; prostate cancer 1. Introduction Splicing of mRNA precursors (pre-mRNAs) is an essential mechanism in the posttranscriptional regulation of gene expression.