Improved Detection of Gene Fusions by Applying Statistical Methods Reveals New Oncogenic RNA Cancer Drivers
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Regulation and Dysregulation of Chromosome Structure in Cancer
Regulation and Dysregulation of Chromosome Structure in Cancer The MIT Faculty has made this article openly available. Please share how this access benefits you. Your story matters. Citation Hnisz, Denes et al. “Regulation and Dysregulation of Chromosome Structure in Cancer.” Annual Review of Cancer Biology 2, 1 (March 2018): 21–40 © 2018 Annual Reviews As Published https://doi.org/10.1146/annurev-cancerbio-030617-050134 Version Author's final manuscript Citable link http://hdl.handle.net/1721.1/117286 Terms of Use Creative Commons Attribution-Noncommercial-Share Alike Detailed Terms http://creativecommons.org/licenses/by-nc-sa/4.0/ Regulation and dysregulation of chromosome structure in cancer Denes Hnisz1*, Jurian Schuijers1, Charles H. Li1,2, Richard A. Young1,2* 1 Whitehead Institute for Biomedical Research, 455 Main Street, Cambridge, MA 02142, USA 2 Department of Biology, Massachusetts Institute of Technology, Cambridge, MA 02139, USA * Corresponding authors Corresponding Authors: Denes Hnisz Whitehead Institute for Biomedical Research 455 Main Street Cambridge, MA 02142 Tel: (617) 258-7181 Fax: (617) 258-0376 [email protected] Richard A. Young Whitehead Institute for Biomedical Research 455 Main Street Cambridge, MA 02142 Tel: (617) 258-5218 Fax: (617) 258-0376 [email protected] 1 Summary Cancer arises from genetic alterations that produce dysregulated gene expression programs. Normal gene regulation occurs in the context of chromosome loop structures called insulated neighborhoods, and recent studies have shown that these structures are altered and can contribute to oncogene dysregulation in various cancer cells. We review here the types of genetic and epigenetic alterations that influence neighborhood structures and contribute to gene dysregulation in cancer, present models for insulated neighborhoods associated with the most prominent human oncogenes, and discuss how such models may lead to further advances in cancer diagnosis and therapy. -

Universidade Estadual De Campinas Instituto De Biologia
UNIVERSIDADE ESTADUAL DE CAMPINAS INSTITUTO DE BIOLOGIA VERÔNICA APARECIDA MONTEIRO SAIA CEREDA O PROTEOMA DO CORPO CALOSO DA ESQUIZOFRENIA THE PROTEOME OF THE CORPUS CALLOSUM IN SCHIZOPHRENIA CAMPINAS 2016 1 VERÔNICA APARECIDA MONTEIRO SAIA CEREDA O PROTEOMA DO CORPO CALOSO DA ESQUIZOFRENIA THE PROTEOME OF THE CORPUS CALLOSUM IN SCHIZOPHRENIA Dissertação apresentada ao Instituto de Biologia da Universidade Estadual de Campinas como parte dos requisitos exigidos para a obtenção do Título de Mestra em Biologia Funcional e Molecular na área de concentração de Bioquímica. Dissertation presented to the Institute of Biology of the University of Campinas in partial fulfillment of the requirements for the degree of Master in Functional and Molecular Biology, in the area of Biochemistry. ESTE ARQUIVO DIGITAL CORRESPONDE À VERSÃO FINAL DA DISSERTAÇÃO DEFENDIDA PELA ALUNA VERÔNICA APARECIDA MONTEIRO SAIA CEREDA E ORIENTADA PELO DANIEL MARTINS-DE-SOUZA. Orientador: Daniel Martins-de-Souza CAMPINAS 2016 2 Agência(s) de fomento e nº(s) de processo(s): CNPq, 151787/2F2014-0 Ficha catalográfica Universidade Estadual de Campinas Biblioteca do Instituto de Biologia Mara Janaina de Oliveira - CRB 8/6972 Saia-Cereda, Verônica Aparecida Monteiro, 1988- Sa21p O proteoma do corpo caloso da esquizofrenia / Verônica Aparecida Monteiro Saia Cereda. – Campinas, SP : [s.n.], 2016. Orientador: Daniel Martins de Souza. Dissertação (mestrado) – Universidade Estadual de Campinas, Instituto de Biologia. 1. Esquizofrenia. 2. Espectrometria de massas. 3. Corpo caloso. -

Sequencing-Based Computational Methods for Identifying Impactful Genomic Alterations in Cancers
Sequencing-based computational methods for identifying impactful genomic alterations in cancers by Isaac Charles Joseph A dissertation submitted in partial satisfaction of the requirements for the degree of Doctor of Philosophy in Computational Biology in the Graduate Division of the University of California, Berkeley Committee in charge: Professor Lior Pachter, Chair Professor Joseph Costello Associate Professor Haiyan Huang Professor Anthony Joseph Fall 2016 Sequencing-based computational methods for identifying impactful genomic alterations in cancers Copyright 2016 by Isaac Charles Joseph 1 Abstract Sequencing-based computational methods for identifying impactful genomic alterations in cancers by Isaac Charles Joseph Doctor of Philosophy in Computational Biology University of California, Berkeley Professor Lior Pachter, Chair Recent advances in collecting sequencing data from tumors is promising for both immediate individual patient treatment and investigation of cancer mechanisms. A resultant central goal is identifying changes in tumors that are impactful towards these ends. Here, we develop two tools to identify impactful changes at different levels. We develop both methods in the context of gliomas, a common form of brain cancer. Firstly, we develop and assess a tool for assessing the impact of fusion genes, a type of common mutation using RNA-sequencing data. We validate the tool by working with collaborators in The Cancer Genome Atlas. Secondly, we develop a tool for an overall assessment of patient outcome by integrating data from diverse sequencing platforms. We validate this tool using simulation, data from consortiums, and collaborators at UCSF. i To those that suffer It is you that have convinced me that there is something still worth fighting for in this world. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

NICU Gene List Generator.Xlsx
Neonatal Crisis Sequencing Panel Gene List Genes: A2ML1 - B3GLCT A2ML1 ADAMTS9 ALG1 ARHGEF15 AAAS ADAMTSL2 ALG11 ARHGEF9 AARS1 ADAR ALG12 ARID1A AARS2 ADARB1 ALG13 ARID1B ABAT ADCY6 ALG14 ARID2 ABCA12 ADD3 ALG2 ARL13B ABCA3 ADGRG1 ALG3 ARL6 ABCA4 ADGRV1 ALG6 ARMC9 ABCB11 ADK ALG8 ARPC1B ABCB4 ADNP ALG9 ARSA ABCC6 ADPRS ALK ARSL ABCC8 ADSL ALMS1 ARX ABCC9 AEBP1 ALOX12B ASAH1 ABCD1 AFF3 ALOXE3 ASCC1 ABCD3 AFF4 ALPK3 ASH1L ABCD4 AFG3L2 ALPL ASL ABHD5 AGA ALS2 ASNS ACAD8 AGK ALX3 ASPA ACAD9 AGL ALX4 ASPM ACADM AGPS AMELX ASS1 ACADS AGRN AMER1 ASXL1 ACADSB AGT AMH ASXL3 ACADVL AGTPBP1 AMHR2 ATAD1 ACAN AGTR1 AMN ATL1 ACAT1 AGXT AMPD2 ATM ACE AHCY AMT ATP1A1 ACO2 AHDC1 ANK1 ATP1A2 ACOX1 AHI1 ANK2 ATP1A3 ACP5 AIFM1 ANKH ATP2A1 ACSF3 AIMP1 ANKLE2 ATP5F1A ACTA1 AIMP2 ANKRD11 ATP5F1D ACTA2 AIRE ANKRD26 ATP5F1E ACTB AKAP9 ANTXR2 ATP6V0A2 ACTC1 AKR1D1 AP1S2 ATP6V1B1 ACTG1 AKT2 AP2S1 ATP7A ACTG2 AKT3 AP3B1 ATP8A2 ACTL6B ALAS2 AP3B2 ATP8B1 ACTN1 ALB AP4B1 ATPAF2 ACTN2 ALDH18A1 AP4M1 ATR ACTN4 ALDH1A3 AP4S1 ATRX ACVR1 ALDH3A2 APC AUH ACVRL1 ALDH4A1 APTX AVPR2 ACY1 ALDH5A1 AR B3GALNT2 ADA ALDH6A1 ARFGEF2 B3GALT6 ADAMTS13 ALDH7A1 ARG1 B3GAT3 ADAMTS2 ALDOB ARHGAP31 B3GLCT Updated: 03/15/2021; v.3.6 1 Neonatal Crisis Sequencing Panel Gene List Genes: B4GALT1 - COL11A2 B4GALT1 C1QBP CD3G CHKB B4GALT7 C3 CD40LG CHMP1A B4GAT1 CA2 CD59 CHRNA1 B9D1 CA5A CD70 CHRNB1 B9D2 CACNA1A CD96 CHRND BAAT CACNA1C CDAN1 CHRNE BBIP1 CACNA1D CDC42 CHRNG BBS1 CACNA1E CDH1 CHST14 BBS10 CACNA1F CDH2 CHST3 BBS12 CACNA1G CDK10 CHUK BBS2 CACNA2D2 CDK13 CILK1 BBS4 CACNB2 CDK5RAP2 -

Characterization of a 7.6-Mb Germline Deletion Encompassing the NF1 Locus and About a Hundred Genes in an NF1 Contiguous Gene Syndrome Patient
European Journal of Human Genetics (2008) 16, 1459–1466 & 2008 Macmillan Publishers Limited All rights reserved 1018-4813/08 $32.00 www.nature.com/ejhg ARTICLE Characterization of a 7.6-Mb germline deletion encompassing the NF1 locus and about a hundred genes in an NF1 contiguous gene syndrome patient Eric Pasmant*,1,2, Aure´lie de Saint-Trivier2, Ingrid Laurendeau1, Anne Dieux-Coeslier3, Be´atrice Parfait1,2, Michel Vidaud1,2, Dominique Vidaud1,2 and Ivan Bie`che1,2 1UMR745 INSERM, Universite´ Paris Descartes, Faculte´ des Sciences Pharmaceutiques et Biologiques, Paris, France; 2Service de Biochimie et de Ge´ne´tique Mole´culaire, Hoˆpital Beaujon AP-HP, Clichy, France; 3Service de Ge´ne´tique Clinique, Hoˆpital Jeanne de Flandre, Lille, France We describe a large germline deletion removing the NF1 locus, identified by heterozygosity mapping based on microsatellite markers, in an 8-year-old French girl with a particularly severe NF1 contiguous gene syndrome. We used gene-dose mapping with sequence-tagged site real-time PCR to locate the deletion end points, which were precisely characterized by means of long-range PCR and nucleotide sequencing. The deletion is located on chromosome arm 17q and is exactly 7 586 986 bp long. It encompasses the entire NF1 locus and about 100 other genes, including numerous chemokine genes, an attractive in silico-selected cerebrally expressed candidate gene (designated NUFIP2, for nuclear fragile X mental retardation protein interacting protein 2; NM_020772) and four microRNA genes. Interestingly, the centromeric breakpoint is located in intron 4 of the PIPOX gene (pipecolic acid oxidase; NM_016518) and the telomeric breakpoint in intron 5 of the GGNBP2 gene (gametogenetin binding protein 2; NM_024835) coding a transcription factor. -
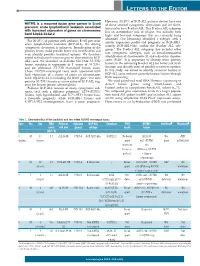
NUTM1 Is a Recurrent Fusion Gene Partner in B-Cell Precursor Acute
LETTERS TO THE EDITOR However, 20-25% of BCP-ALL patients do not have one NUTM1 is a recurrent fusion gene partner in B-cell of these sentinel cytogenetic aberrations and are there- precursor acute lymphoblastic leukemia associated fore said to have B-other ALL. This B-other ALL subgroup with increased expression of genes on chromosome has an intermediate risk of relapse, but includes both band 10p12.31-12.2 high- and low-risk subgroups that are currently being identified. Our laboratory identified a subtype with a For 20-25% of patients with pediatric B-cell precursor similar expression profile and prognosis as BCR-ABL1, acute lymphoblastic leukemia (BCP-ALL), the driving namely BCR-ABL1-like, within the B-other ALL sub- cytogenetic aberration is unknown. Identification of the group.2 The B-other ALL subgroup also includes other primary lesion could provide better risk stratification and rare cytogenetic subtypes, such as intrachromosomal even identify possible treatment options. We therefore amplification of chromosome 21 and a dicentric chromo- aimed to find novel recurrent genetic aberrations in BCP- 1 ALL cases. We identified an in-frame SLC12A6-NUTM1 some (9;20). It is important to identify more primary fusion, resulting in expression of 3’ exons of NUTM1, lesions in the remaining B-other ALL for better risk strat- and six additional NUTM1-rearranged fusion cases. ification and identification of possible treatment options. These NUTM1-rearranged cases were associated with In this study, we aimed to identify recurrent fusions in high expression of a cluster of genes on chromosome BCP-ALL cases without currently known lesions through band 10p12.31-12.2, including the BMI1 gene. -

4 Understanding the Role of GNA13 Deregulation in Lymphomagenesis
Integrative Genomics Reveals a Role for GNA13 in Lymphomagenesis by Adrienne Greenough University Program in Genetics and Genomics Duke University Approved: ___________________________ Sandeep Dave, Supervisor ___________________________ Fred Dietrich ___________________________ Jack Keene ___________________________ Yuan Zhuang Dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the University Program in Genetics and Genomics in the Graduate School of Duke University 2014 i v ABSTRACT Integrative Genomics Reveals a Role for GNA13 in Lymphomagenesis by Adrienne Greenough University Program in Genetics and Genomics Duke University Approved: ___________________________ Sandeep Dave, Supervisor ___________________________ Fred Dietrich ___________________________ Jack Keene ___________________________ Yuan Zhuang An abstract of a dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the University Program in Genetics and Genomics in the Graduate School of Duke University 2014 Copyright by Adrienne Greenough 2014 Abstract Lymphomas comprise a diverse group of malignancies derived from immune cells. High throughput sequencing has recently emerged as a powerful and versatile method for analysis of the cancer genome and transcriptome. As these data continue to emerge, the crucial work lies in sorting through the wealth of information to hone in on the critical aspects that will give us a better understanding of biology and new insight for how to treat disease. Finding the important signals within these large data sets is one of the major challenges of next generation sequencing. In this dissertation, I have developed several complementary strategies to describe the genetic underpinnings of lymphomas. I begin with developing a better method for RNA sequencing that enables strand-specific total RNA sequencing and alternative splicing profiling in the same analysis. -

Multi-Functionality of Proteins Involved in GPCR and G Protein Signaling: Making Sense of Structure–Function Continuum with In
Cellular and Molecular Life Sciences (2019) 76:4461–4492 https://doi.org/10.1007/s00018-019-03276-1 Cellular andMolecular Life Sciences REVIEW Multi‑functionality of proteins involved in GPCR and G protein signaling: making sense of structure–function continuum with intrinsic disorder‑based proteoforms Alexander V. Fonin1 · April L. Darling2 · Irina M. Kuznetsova1 · Konstantin K. Turoverov1,3 · Vladimir N. Uversky2,4 Received: 5 August 2019 / Revised: 5 August 2019 / Accepted: 12 August 2019 / Published online: 19 August 2019 © Springer Nature Switzerland AG 2019 Abstract GPCR–G protein signaling system recognizes a multitude of extracellular ligands and triggers a variety of intracellular signal- ing cascades in response. In humans, this system includes more than 800 various GPCRs and a large set of heterotrimeric G proteins. Complexity of this system goes far beyond a multitude of pair-wise ligand–GPCR and GPCR–G protein interactions. In fact, one GPCR can recognize more than one extracellular signal and interact with more than one G protein. Furthermore, one ligand can activate more than one GPCR, and multiple GPCRs can couple to the same G protein. This defnes an intricate multifunctionality of this important signaling system. Here, we show that the multifunctionality of GPCR–G protein system represents an illustrative example of the protein structure–function continuum, where structures of the involved proteins represent a complex mosaic of diferently folded regions (foldons, non-foldons, unfoldons, semi-foldons, and inducible foldons). The functionality of resulting highly dynamic conformational ensembles is fne-tuned by various post-translational modifcations and alternative splicing, and such ensembles can undergo dramatic changes at interaction with their specifc partners. -

Transposable Elements in Human Cancers by Genome-Wide EST Alignment
Genes Genet. Syst. (2007) 82, p. 145–156 Transposable elements in human cancers by genome-wide EST alignment Dae-Soo Kim1, Jae-Won Huh2 and Heui-Soo Kim1,2* 1PBBRC, Interdisciplinary Research Program of Bioinformatics, Pusan National University, Busan 609-735, Republic of Korea 2Division of Biological Sciences, College of Natural Sciences, Pusan National University, Busan 609-735, Republic of Korea (Received 24 November 2006, accepted 23 January 2007) Transposable elements may affect coding sequences, splicing patterns, and tran- scriptional regulation of human genes. Particles of the transposable elements have been detected in several tissues and tumors. Here, we report genome-wide analysis of gene expression regulated by transposable elements in human cancers. We adopted an analysis pipeline for screening methods to detect cancer- specific expression from expressed human sequences. We developed a database (TECESdb) for understanding the mechanism of cancer development in relation to transposable elements. A total of 999 genes fused with transposable elements were found to be cancer-related in our analysis of the EST database. According to GO (Gene Ontology) analysis, the majority of the 999 cancer-specific genes have functional association with gene receptor, DNA binding, and kinase activity. Our data could contribute greatly to our understanding of human cancers in relation to transposable elements. Key words: Transposable elements, Cancer, Fusion gene, Bioinformatics, EST also appeared in open-reading frames of functional INTRODUCTION human genes (Yulug et al., 1995; Makalowski et al., 1999; The human genome is estimated to be composed of 45% Nekrutenko and Li, 2001; Huh et al., 2006). transposable elements (International Human Genome The L1 5’UTR element is known to have an antisense Sequencing Consortium 2001). -

Improved Detection of Gene Fusions by Applying Statistical Methods Reveals New Oncogenic RNA Cancer Drivers
bioRxiv preprint doi: https://doi.org/10.1101/659078; this version posted June 3, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Improved detection of gene fusions by applying statistical methods reveals new oncogenic RNA cancer drivers Roozbeh Dehghannasiri1, Donald Eric Freeman1,2, Milos Jordanski3, Gillian L. Hsieh1, Ana Damljanovic4, Erik Lehnert4, Julia Salzman1,2,5* Author affiliation 1Department of Biochemistry, Stanford University, Stanford, CA 94305 2Department of Biomedical Data Science, Stanford University, Stanford, CA 94305 3Department of Computer Science, University of Belgrade, Belgrade, Serbia 4Seven Bridges Genomics, Cambridge, MA 02142 5Stanford Cancer Institute, Stanford, CA 94305 *Corresponding author [email protected] Short Abstract: The extent to which gene fusions function as drivers of cancer remains a critical open question. Current algorithms do not sufficiently identify false-positive fusions arising during library preparation, sequencing, and alignment. Here, we introduce a new algorithm, DEEPEST, that uses statistical modeling to minimize false-positives while increasing the sensitivity of fusion detection. In 9,946 tumor RNA-sequencing datasets from The Cancer Genome Atlas (TCGA) across 33 tumor types, DEEPEST identifies 31,007 fusions, 30% more than identified by other methods, while calling ten-fold fewer false-positive fusions in non-transformed human tissues. We leverage the increased precision of DEEPEST to discover new cancer biology. For example, 888 new candidate oncogenes are identified based on over-representation in DEEPEST-Fusion calls, and 1,078 previously unreported fusions involving long intergenic noncoding RNAs partners, demonstrating a previously unappreciated prevalence and potential for function. -

EWSR1 Gene EWS RNA Binding Protein 1
EWSR1 gene EWS RNA binding protein 1 Normal Function The EWSR1 gene provides instructions for making the EWS protein, whose function is not completely understood. The EWS protein has two regions that contribute to its function. One region, the transcriptional activation domain, allows the EWS protein to turn on (activate) the first step in the production of proteins from genes (transcription). The other region, the RNA-binding domain, allows the EWS protein to attach (bind) to the genetic blueprint for proteins called RNA. The EWS protein may be involved in piecing together this blueprint. Some studies suggest that the RNA-binding domain is able to block (inhibit) the activity of the transcriptional activation domain, and thus regulate the function of the EWS protein. Health Conditions Related to Genetic Changes Ewing sarcoma Mutations involving the EWSR1 gene can cause a type of cancerous tumor known as Ewing sarcoma. These tumors develop in bones or soft tissues, such as nerves and cartilage. There are several types of Ewing sarcoma, including Ewing sarcoma of bone, extraosseous Ewing sarcoma, peripheral primitive neuroectodermal tumor, and Askin tumor. The mutations that cause these tumors are acquired during a person's lifetime and are present only in the tumor cells. This type of genetic change, called a somatic mutation, is not inherited. The most common mutation that causes Ewing sarcoma is a rearrangement (translocation) of genetic material between chromosome 22 and chromosome 11. This translocation, written as t(11;22), fuses part of the EWSR1 gene on chromosome 22 with part of another gene on chromosome 11 called FLI1, creating an EWSR1/FLI1 fusion gene.