Gαo (GNAO1) Encephalopathies: Plasma Membrane Vs. Golgi Functions
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

NICU Gene List Generator.Xlsx
Neonatal Crisis Sequencing Panel Gene List Genes: A2ML1 - B3GLCT A2ML1 ADAMTS9 ALG1 ARHGEF15 AAAS ADAMTSL2 ALG11 ARHGEF9 AARS1 ADAR ALG12 ARID1A AARS2 ADARB1 ALG13 ARID1B ABAT ADCY6 ALG14 ARID2 ABCA12 ADD3 ALG2 ARL13B ABCA3 ADGRG1 ALG3 ARL6 ABCA4 ADGRV1 ALG6 ARMC9 ABCB11 ADK ALG8 ARPC1B ABCB4 ADNP ALG9 ARSA ABCC6 ADPRS ALK ARSL ABCC8 ADSL ALMS1 ARX ABCC9 AEBP1 ALOX12B ASAH1 ABCD1 AFF3 ALOXE3 ASCC1 ABCD3 AFF4 ALPK3 ASH1L ABCD4 AFG3L2 ALPL ASL ABHD5 AGA ALS2 ASNS ACAD8 AGK ALX3 ASPA ACAD9 AGL ALX4 ASPM ACADM AGPS AMELX ASS1 ACADS AGRN AMER1 ASXL1 ACADSB AGT AMH ASXL3 ACADVL AGTPBP1 AMHR2 ATAD1 ACAN AGTR1 AMN ATL1 ACAT1 AGXT AMPD2 ATM ACE AHCY AMT ATP1A1 ACO2 AHDC1 ANK1 ATP1A2 ACOX1 AHI1 ANK2 ATP1A3 ACP5 AIFM1 ANKH ATP2A1 ACSF3 AIMP1 ANKLE2 ATP5F1A ACTA1 AIMP2 ANKRD11 ATP5F1D ACTA2 AIRE ANKRD26 ATP5F1E ACTB AKAP9 ANTXR2 ATP6V0A2 ACTC1 AKR1D1 AP1S2 ATP6V1B1 ACTG1 AKT2 AP2S1 ATP7A ACTG2 AKT3 AP3B1 ATP8A2 ACTL6B ALAS2 AP3B2 ATP8B1 ACTN1 ALB AP4B1 ATPAF2 ACTN2 ALDH18A1 AP4M1 ATR ACTN4 ALDH1A3 AP4S1 ATRX ACVR1 ALDH3A2 APC AUH ACVRL1 ALDH4A1 APTX AVPR2 ACY1 ALDH5A1 AR B3GALNT2 ADA ALDH6A1 ARFGEF2 B3GALT6 ADAMTS13 ALDH7A1 ARG1 B3GAT3 ADAMTS2 ALDOB ARHGAP31 B3GLCT Updated: 03/15/2021; v.3.6 1 Neonatal Crisis Sequencing Panel Gene List Genes: B4GALT1 - COL11A2 B4GALT1 C1QBP CD3G CHKB B4GALT7 C3 CD40LG CHMP1A B4GAT1 CA2 CD59 CHRNA1 B9D1 CA5A CD70 CHRNB1 B9D2 CACNA1A CD96 CHRND BAAT CACNA1C CDAN1 CHRNE BBIP1 CACNA1D CDC42 CHRNG BBS1 CACNA1E CDH1 CHST14 BBS10 CACNA1F CDH2 CHST3 BBS12 CACNA1G CDK10 CHUK BBS2 CACNA2D2 CDK13 CILK1 BBS4 CACNB2 CDK5RAP2 -

4 Understanding the Role of GNA13 Deregulation in Lymphomagenesis
Integrative Genomics Reveals a Role for GNA13 in Lymphomagenesis by Adrienne Greenough University Program in Genetics and Genomics Duke University Approved: ___________________________ Sandeep Dave, Supervisor ___________________________ Fred Dietrich ___________________________ Jack Keene ___________________________ Yuan Zhuang Dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the University Program in Genetics and Genomics in the Graduate School of Duke University 2014 i v ABSTRACT Integrative Genomics Reveals a Role for GNA13 in Lymphomagenesis by Adrienne Greenough University Program in Genetics and Genomics Duke University Approved: ___________________________ Sandeep Dave, Supervisor ___________________________ Fred Dietrich ___________________________ Jack Keene ___________________________ Yuan Zhuang An abstract of a dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the University Program in Genetics and Genomics in the Graduate School of Duke University 2014 Copyright by Adrienne Greenough 2014 Abstract Lymphomas comprise a diverse group of malignancies derived from immune cells. High throughput sequencing has recently emerged as a powerful and versatile method for analysis of the cancer genome and transcriptome. As these data continue to emerge, the crucial work lies in sorting through the wealth of information to hone in on the critical aspects that will give us a better understanding of biology and new insight for how to treat disease. Finding the important signals within these large data sets is one of the major challenges of next generation sequencing. In this dissertation, I have developed several complementary strategies to describe the genetic underpinnings of lymphomas. I begin with developing a better method for RNA sequencing that enables strand-specific total RNA sequencing and alternative splicing profiling in the same analysis. -

Multi-Functionality of Proteins Involved in GPCR and G Protein Signaling: Making Sense of Structure–Function Continuum with In
Cellular and Molecular Life Sciences (2019) 76:4461–4492 https://doi.org/10.1007/s00018-019-03276-1 Cellular andMolecular Life Sciences REVIEW Multi‑functionality of proteins involved in GPCR and G protein signaling: making sense of structure–function continuum with intrinsic disorder‑based proteoforms Alexander V. Fonin1 · April L. Darling2 · Irina M. Kuznetsova1 · Konstantin K. Turoverov1,3 · Vladimir N. Uversky2,4 Received: 5 August 2019 / Revised: 5 August 2019 / Accepted: 12 August 2019 / Published online: 19 August 2019 © Springer Nature Switzerland AG 2019 Abstract GPCR–G protein signaling system recognizes a multitude of extracellular ligands and triggers a variety of intracellular signal- ing cascades in response. In humans, this system includes more than 800 various GPCRs and a large set of heterotrimeric G proteins. Complexity of this system goes far beyond a multitude of pair-wise ligand–GPCR and GPCR–G protein interactions. In fact, one GPCR can recognize more than one extracellular signal and interact with more than one G protein. Furthermore, one ligand can activate more than one GPCR, and multiple GPCRs can couple to the same G protein. This defnes an intricate multifunctionality of this important signaling system. Here, we show that the multifunctionality of GPCR–G protein system represents an illustrative example of the protein structure–function continuum, where structures of the involved proteins represent a complex mosaic of diferently folded regions (foldons, non-foldons, unfoldons, semi-foldons, and inducible foldons). The functionality of resulting highly dynamic conformational ensembles is fne-tuned by various post-translational modifcations and alternative splicing, and such ensembles can undergo dramatic changes at interaction with their specifc partners. -

Methylation of Leukocyte DNA and Ovarian Cancer
Fridley et al. BMC Medical Genomics 2014, 7:21 http://www.biomedcentral.com/1755-8794/7/21 RESEARCH ARTICLE Open Access Methylation of leukocyte DNA and ovarian cancer: relationships with disease status and outcome Brooke L Fridley1*, Sebastian M Armasu2, Mine S Cicek2, Melissa C Larson2, Chen Wang2, Stacey J Winham2, Kimberly R Kalli3, Devin C Koestler1,4, David N Rider2, Viji Shridhar5, Janet E Olson2, Julie M Cunningham5 and Ellen L Goode2 Abstract Background: Genome-wide interrogation of DNA methylation (DNAm) in blood-derived leukocytes has become feasible with the advent of CpG genotyping arrays. In epithelial ovarian cancer (EOC), one report found substantial DNAm differences between cases and controls; however, many of these disease-associated CpGs were attributed to differences in white blood cell type distributions. Methods: We examined blood-based DNAm in 336 EOC cases and 398 controls; we included only high-quality CpG loci that did not show evidence of association with white blood cell type distributions to evaluate association with case status and overall survival. Results: Of 13,816 CpGs, no significant associations were observed with survival, although eight CpGs associated with survival at p < 10−3, including methylation within a CpG island located in the promoter region of GABRE (p = 5.38 x 10−5, HR = 0.95). In contrast, 53 CpG methylation sites were significantly associated with EOC risk (p <5 x10−6). The top association was observed for the methylation probe cg04834572 located approximately 315 kb upstream of DUSP13 (p = 1.6 x10−14). Other disease-associated CpGs included those near or within HHIP (cg14580567; p =5.6x10−11), HDAC3 (cg10414058; p = 6.3x10−12), and SCR (cg05498681; p = 4.8x10−7). -

Diallyl Disulfide Inhibits the Proliferation of HT-29 Human Colon Cancer Cells by Inducing Differentially Expressed Genes
MOLECULAR MEDICINE REPORTS 4: 553-559, 2011 Diallyl disulfide inhibits the proliferation of HT-29 human colon cancer cells by inducing differentially expressed genes YOU-SHENG HUANG1,2, NA XIE1,2, QI SU1, JIAN SU1, CHEN HUANG1 and QIAN-JIN LIAO1 1Cancer Research Institute, University of South China, Hengyang, Hunan 421001; 2Department of Pathology, Hainan Medical University, Haikou, Hainan 571101, P.R. China Received November 22, 2010; Accepted February 28, 2011 DOI: 10.3892/mmr.2011.453 Abstract. Diallyl disulfide (DADS), a sulfur compound Introduction derived from garlic, has been shown to have protective effects against colon carcinogenesis in several studies performed in Colon cancer is one of the major causes of cancer death rodent models. However, its molecular mechanism of action worldwide (1). An understanding of the mechanisms involved remains unclear. This study was designed to confirm the anti- in the occurrence and development of colon cancer would aid proliferative activity of DADS and to screen for differentially in its therapy. Epidemiological investigations have provided expressed genes induced by DADS in human colon cancer cells compelling evidence that environmental factors are modifiers with the aim of exploring its possible anticancer mechanisms. in colon cancer (1-3); diet has also been shown to be an impor- The anti-proliferative capability of DADS in the HT-29 human tant determinant of cancer risk and tumor behavior (3-5). colon cancer cells was analyzed by MTT assays and flow Garlic consumption is very popular all over the world. cytometry. The differences in gene expression between DADS- Epidemiological studies have shown an inverse correlation treated (experimental group) and untreated (control group) between the consumption of garlic and colon cancer in certain HT-29 cells were identified using two-directional suppression areas (6). -

Mutations in G Protein Β Subunits Promote Transformation and Kinase Inhibitor Resistance
Mutations in G protein β subunits promote transformation and kinase inhibitor resistance The Harvard community has made this article openly available. Please share how this access benefits you. Your story matters Citation Yoda, Akinori, Guillaume Adelmant, Jerome Tamburini, Bjoern Chapuy, Nobuaki Shindoh, Yuka Yoda, Oliver Weigert, et al. 2014. “Mutations in G Protein β Subunits Promote Transformation and Kinase Inhibitor Resistance.” Nature Medicine 21 (1) (December 8): 71–75. doi:10.1038/nm.3751. Published Version doi:10.1038/nm.3751 Citable link http://nrs.harvard.edu/urn-3:HUL.InstRepos:33751019 Terms of Use This article was downloaded from Harvard University’s DASH repository, and is made available under the terms and conditions applicable to Open Access Policy Articles, as set forth at http:// nrs.harvard.edu/urn-3:HUL.InstRepos:dash.current.terms-of- use#OAP HHS Public Access Author manuscript Author Manuscript Author ManuscriptNat Med Author Manuscript. Author manuscript; Author Manuscript available in PMC 2015 July 01. Published in final edited form as: Nat Med. 2015 January ; 21(1): 71–75. doi:10.1038/nm.3751. Mutations in G protein beta subunits promote transformation and kinase inhibitor resistance Akinori Yoda1, Guillaume Adelmant2, Jerome Tamburini1, Bjoern Chapuy1, Nobuaki Shindoh1,3, Yuka Yoda1, Oliver Weigert4, Nadja Kopp1, Shuo-Chieh Wu1, Sunhee S. Kim1, Huiyun Liu1, Trevor Tivey1, Amanda L. Christie1, Kutlu G. Elpek5,6, Joseph Card2, Kira Gritsman1, Jason Gotlib7, Michael W. Deininger8, Hideki Makishima9, Shannon J. Turley5, Nathalie Javidi-Sharifi10, Jaroslaw P. Maciejewski9, Siddhartha Jaiswal11,12, Benjamin L. Ebert12,13, Scott J. Rodig14, Jeffrey W. Tyner10, Jarrod A. -

TDP-43 Dysfunction Restricts Dendritic Complexity by Inhibiting CREB
bioRxiv preprint doi: https://doi.org/10.1101/2019.12.12.874735; this version posted December 13, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. TDP-43 dysfunction restricts dendritic complexity by inhibiting CREB activation and altering gene expression Josiah J. Herzog1,2, Mugdha Deshpande1,2, Weijin Xu2,3, Reazur Rahman2,3, Hannah Suib2,3, Michael Rosbash2,3,4, Avital A. Rodal3,4,5, Suzanne Paradis4,6 1Equal Contribution 2Department of Biology, Brandeis University, Waltham, Massachusetts 02454 3Howard Hughes Medical Institute, Brandeis University, Waltham, MA 02454 4Volen Center for Complex Systems, Brandeis University, Waltham, MA 02454 5Rosenstiel Basic Medical Sciences Research Center, Brandeis University, Waltham, MA 02454 6Department of Biology, Brandeis University, Waltham, Massachusetts 02454; [email protected] 1 bioRxiv preprint doi: https://doi.org/10.1101/2019.12.12.874735; this version posted December 13, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. Abstract Amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) are two related neurodegenerative diseases that present with similar TDP-43 pathology in patient tissue. TDP-43 is an RNA-binding protein and forms aggregates in neurons of ALS and FTD patients as well as in a subset of patients diagnosed with other neurodegenerative diseases. -

Doxazosin Induces Activation of GADD153 and Cleavage of Focal
Cardiovascular Research 71 (2006) 118 – 128 www.elsevier.com/locate/cardiores Doxazosin induces activation of GADD153 and cleavage of focal adhesion kinase in cardiomyocytes en route to apoptosis Downloaded from https://academic.oup.com/cardiovascres/article/71/1/118/269227 by guest on 01 October 2021 Sonia Eiras, Patricia Ferna´ndez, Roberto Pin˜eiro, Marı´a Jesu´s Iglesias, Jose´ Ramo´n Gonza´lez-Juanatey, Francisca Lago * Unidad de Investigacio´n del Servicio de Cardiologı´a, Hospital Clı´nico Universitario, Santiago de Compostela, Spain Received 14 October 2005; received in revised form 3 March 2006; accepted 17 March 2006 Available online 24 March 2006 Time for primary review 31 days Abstract Objective: The a1-adrenoreceptor blocker doxazosin, which in the ALLHAT trial was associated with a greater risk of heart failure than the diuretic chlorthalidone, induces the apoptosis of human and murine cardiomyocytes regardless of a1-adrenoreceptor blockade. We aimed to throw light on the mechanism of this process. Methods: Murine cardiomyocytes (HL-1) and primary cultures of human and neonatal rat cardiomyocytes were treated with 25 Amol/L doxazosin for between 0.5 and 48 h. cDNA microarray analysis, real-time RT-PCR, and Western blotting were performed to detect possible changes in gene expression and/or activation of proteins that could be involved in doxazosin-induced apoptosis. Results: Microarray analysis revealed changes in the expression of genes directly involved in the apoptotic end-stage of the cellular response to endoplasmic reticulum (ER) stress. Doxazosin considerably increased transcription and translation of gadd153, C/epbb, and DOC-1 in cardiomyocytes as well as translocation of GADD153 to the nucleus, phosphorylation of p38 MAPK (a GADD153 activator), and the initial phosphorylation and subsequent cleavage of focal adhesion kinase (FAK). -

Dema and Faust Et Al., Suppl. Material 2020.02.03
Supplementary Materials Cyclin-dependent kinase 18 controls trafficking of aquaporin-2 and its abundance through ubiquitin ligase STUB1, which functions as an AKAP Dema Alessandro1,2¶, Dörte Faust1¶, Katina Lazarow3, Marc Wippich3, Martin Neuenschwander3, Kerstin Zühlke1, Andrea Geelhaar1, Tamara Pallien1, Eileen Hallscheidt1, Jenny Eichhorst3, Burkhard Wiesner3, Hana Černecká1, Oliver Popp1, Philipp Mertins1, Gunnar Dittmar1, Jens Peter von Kries3, Enno Klussmann1,4* ¶These authors contributed equally to this work 1Max Delbrück Center for Molecular Medicine in the Helmholtz Association (MDC), Robert- Rössle-Strasse 10, 13125 Berlin, Germany 2current address: University of California, San Francisco, 513 Parnassus Avenue, CA 94122 USA 3Leibniz-Forschungsinstitut für Molekulare Pharmakologie (FMP), Robert-Rössle-Strasse 10, 13125 Berlin, Germany 4DZHK (German Centre for Cardiovascular Research), Partner Site Berlin, Oudenarder Strasse 16, 13347 Berlin, Germany *Corresponding author Enno Klussmann Max Delbrück Center for Molecular Medicine Berlin in the Helmholtz Association (MDC) Robert-Rössle-Str. 10, 13125 Berlin Germany Tel. +49-30-9406 2596 FAX +49-30-9406 2593 E-mail: [email protected] 1 Content 1. CELL-BASED SCREENING BY AUTOMATED IMMUNOFLUORESCENCE MICROSCOPY 3 1.1 Screening plates 3 1.2 Image analysis using CellProfiler 17 1.4 Identification of siRNA affecting cell viability 18 1.7 Hits 18 2. SUPPLEMENTARY TABLE S4, FIGURES S2-S4 20 2 1. Cell-based screening by automated immunofluorescence microscopy 1.1 Screening plates Table S1. Genes targeted with the Mouse Protein Kinases siRNA sub-library. Genes are sorted by plate and well. Accessions refer to National Center for Biotechnology Information (NCBI, BLA) entries. The siRNAs were arranged on three 384-well microtitre platres. -

UNIVERSITY of CALIFORNIA Los Angeles Identification of a Multisubunit E3 Ubiquitin Ligase Required for Heterotrimeric G-Protein
UNIVERSITY OF CALIFORNIA Los Angeles Identification of a multisubunit E3 ubiquitin ligase required for heterotrimeric G-protein beta-subunit ubiquitination and cAMP signaling A dissertation submitted in partial satisfaction of the requirements for the degree Doctor of Philosophy in Molecular Biology by Brian Daniel Young 2018 © Copyright by Brian Daniel Young 2018 ABSTRACT OF THE DISSERTATION Identification of a multisubunit E3 ubiquitin ligase required for heterotrimeric G-protein beta-subunit ubiquitination and cAMP signaling by Brian Daniel Young Doctor of Philosophy in Molecular Biology University of California, Los Angeles, 2018 Professor James Akira Wohlschlegel, Chair GPCRs are stimulated by extracellular ligands and initiate a range of intracellular signaling events through heterotrimeric G-proteins. Upon activation, G-protein α- subunits (Gα) and the stable βγ-subunit dimer (Gβγ) bind and alter the activity of diverse effectors. These signaling events are fundamental and subject to multiple layers of regulation. In this study, we used an unbiased proteomic mass spectrometry approach to uncover novel regulators of Gβγ. We identified a subfamily of potassium channel tetramerization domain (KCTD) proteins that specifically bind Gβγ. Several KCTD proteins are substrate adaptor proteins for CUL3–RING E3 ubiquitin ligases. Our studies revealed that a KCTD2-KCTD5 hetero-oligomer associates with CUL3 through KCTD5 subunits and recruits Gβγ through both subunits. Using in vitro ubiquitination reactions, we demonstrated that these KCTD proteins promote monoubiquitination of lysine-23 within Gβ1/2. This ubiquitin modification of Gβ1/2 is also observed in human ii cells and is dependent on these substrate adaptor proteins. Because these KCTD proteins bind Gβγ in response to G-protein activation, we investigated their role in GPCR signaling. -
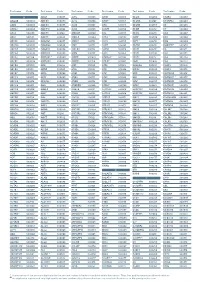
Aagab S00002 Aars S00003 Aars2 S00004 Aass S02483
Test name Code Test name Code Test name Code Test name Code Test name Code Test name Code A ADAR S00053 ALPL S00105 ARSB S00153 BCL10 S02266 C5AR2 S00263 AAGAB S00002 ADCK3 S00054 ALS2 S00106 ARSE * S00154 BCL11A S02167 C5ORF42 S00264 AARS S00003 ADCK4 S00055 ALX3 S00107 ARX S00155 BCL11B S02358 C6 S00265 AARS2 S00004 ADCY10 S02094 ALX4 S00108 ASAH1 S00156 BCOR S00212 C7 S00266 AASS S02483 ADCY3 S02184 AMACR S00109 ASL S00157 BCS1L S00213 C8A S00267 ABAT S02191 ADCY5 S02226 AMELX S02289 ASNS * S02508 BDNF S02509 C8B S00268 ABCA1 S00005 ADGRG1 S00057 AMER1 S00110 ASPA S00158 BDP1 * S00214 C8G S00269 ABCA12 S00006 ADGRG6 S02548 AMH S00111 ASPH S02425 BEAN1 S00215 C8ORF37 S00270 ABCA3 S00007 ADGRV1 S00058 AMHR2 S00112 ASPM S00159 BEST1 S00216 C9 S00271 ABCA4 S00008 ADIPOQ S00059 AMN S00113 ASS1 S00160 BFSP1 S02280 CA2 S00272 ABCA7 S02106 ADIPOR1 * S00060 AMPD1 S02670 ATAD3A * S02196 BFSP2 S00217 CA4 S02303 ABCB11 S00009 ADIPOR2 S00061 AMPD2 S02128 ATCAY S00162 BGN S02633 CA8 S00273 ABCB4 S00010 ADK S02595 AMT S00114 ATF6 S00163 BHLHA9 S00218 CABP2 S00274 ABCB6 S00011 ADNP S02320 ANG S00115 ATIC S02458 BICD2 S00220 CABP4 S00275 ABCB7 S00012 ADSL S00062 ANK1 S00116 ATL1 S00164 BIN1 S00221 CACNA1A S00276 ABCC2 S00013 AFF2 S00063 ANK2 S00117 ATL3 S00165 BLK S00222 CACNA1C * S00277 ABCC6 * S00014 AFG3L2 * S00064 ANKH S00118 ATM S00166 BLM S00223 CACNA1D S00278 ABCC8 S00015 AGA S00065 ANKRD11 * S02140 ATOH7 S02390 BLNK S02281 CACNA1F S00279 ABCC9 S00016 AGBL5 S02452 ANKS6 S00121 ATP13A2 S00168 BLOC1S3 S00224 CACNA1H S00280 ABCD1 * S00017 AGK * -

SUPPLEMENTARY APPENDIX Exome Sequencing Reveals Heterogeneous Clonal Dynamics in Donor Cell Myeloid Neoplasms After Stem Cell Transplantation
SUPPLEMENTARY APPENDIX Exome sequencing reveals heterogeneous clonal dynamics in donor cell myeloid neoplasms after stem cell transplantation Julia Suárez-González, 1,2 Juan Carlos Triviño, 3 Guiomar Bautista, 4 José Antonio García-Marco, 4 Ángela Figuera, 5 Antonio Balas, 6 José Luis Vicario, 6 Francisco José Ortuño, 7 Raúl Teruel, 7 José María Álamo, 8 Diego Carbonell, 2,9 Cristina Andrés-Zayas, 1,2 Nieves Dorado, 2,9 Gabriela Rodríguez-Macías, 9 Mi Kwon, 2,9 José Luis Díez-Martín, 2,9,10 Carolina Martínez-Laperche 2,9* and Ismael Buño 1,2,9,11* on behalf of the Spanish Group for Hematopoietic Transplantation (GETH) 1Genomics Unit, Gregorio Marañón General University Hospital, Gregorio Marañón Health Research Institute (IiSGM), Madrid; 2Gregorio Marañón Health Research Institute (IiSGM), Madrid; 3Sistemas Genómicos, Valencia; 4Department of Hematology, Puerta de Hierro General University Hospital, Madrid; 5Department of Hematology, La Princesa University Hospital, Madrid; 6Department of Histocompatibility, Madrid Blood Centre, Madrid; 7Department of Hematology and Medical Oncology Unit, IMIB-Arrixaca, Morales Meseguer General University Hospital, Murcia; 8Centro Inmunológico de Alicante - CIALAB, Alicante; 9Department of Hematology, Gregorio Marañón General University Hospital, Madrid; 10 Department of Medicine, School of Medicine, Com - plutense University of Madrid, Madrid and 11 Department of Cell Biology, School of Medicine, Complutense University of Madrid, Madrid, Spain *CM-L and IB contributed equally as co-senior authors. Correspondence: