A Meta-Analysis of RNA-Seq Datasets
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Analysis of Gene Expression Data for Gene Ontology
ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION A Thesis Presented to The Graduate Faculty of The University of Akron In Partial Fulfillment of the Requirements for the Degree Master of Science Robert Daniel Macholan May 2011 ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION Robert Daniel Macholan Thesis Approved: Accepted: _______________________________ _______________________________ Advisor Department Chair Dr. Zhong-Hui Duan Dr. Chien-Chung Chan _______________________________ _______________________________ Committee Member Dean of the College Dr. Chien-Chung Chan Dr. Chand K. Midha _______________________________ _______________________________ Committee Member Dean of the Graduate School Dr. Yingcai Xiao Dr. George R. Newkome _______________________________ Date ii ABSTRACT A tremendous increase in genomic data has encouraged biologists to turn to bioinformatics in order to assist in its interpretation and processing. One of the present challenges that need to be overcome in order to understand this data more completely is the development of a reliable method to accurately predict the function of a protein from its genomic information. This study focuses on developing an effective algorithm for protein function prediction. The algorithm is based on proteins that have similar expression patterns. The similarity of the expression data is determined using a novel measure, the slope matrix. The slope matrix introduces a normalized method for the comparison of expression levels throughout a proteome. The algorithm is tested using real microarray gene expression data. Their functions are characterized using gene ontology annotations. The results of the case study indicate the protein function prediction algorithm developed is comparable to the prediction algorithms that are based on the annotations of homologous proteins. -

Trafficking Routes to the Plant Vacuole: Connecting Alternative and Classical Pathways
This is a repository copy of Trafficking routes to the plant vacuole: connecting alternative and classical pathways. White Rose Research Online URL for this paper: http://eprints.whiterose.ac.uk/124374/ Version: Accepted Version Article: Di Sansebastiano, GP, Barozzi, F, Piro, G et al. (2 more authors) (2018) Trafficking routes to the plant vacuole: connecting alternative and classical pathways. Journal of Experimental Botany, 69 (1). pp. 79-90. ISSN 0022-0957 https://doi.org/10.1093/jxb/erx376 © The Author(s) 2017. Published by Oxford University Press on behalf of the Society for Experimental Biology. This is an author produced version of a paper published in Journal of Experimental Botany. Uploaded in accordance with the publisher's self-archiving policy. Reuse Items deposited in White Rose Research Online are protected by copyright, with all rights reserved unless indicated otherwise. They may be downloaded and/or printed for private study, or other acts as permitted by national copyright laws. The publisher or other rights holders may allow further reproduction and re-use of the full text version. This is indicated by the licence information on the White Rose Research Online record for the item. Takedown If you consider content in White Rose Research Online to be in breach of UK law, please notify us by emailing [email protected] including the URL of the record and the reason for the withdrawal request. [email protected] https://eprints.whiterose.ac.uk/ 1 Trafficking routes to the Plant Vacuole: 2 connecting alternative and classical pathways. 3 4 Gian Pietro Di Sansebastiano1*, Fabrizio Barozzi1, Gabriella Piro1, Jurgen Denecke2 and 5 Carine de Marcos Lousa2,3 * 6 1.! DiSTeBA (Dipartimento di Scienze e Tecnologie Biologiche ed Ambientali), 7 University of Salento, Campus ECOTEKNE, 73100 Lecce, Italy; 8 [email protected] and [email protected]. -

Sorting Nexins in Protein Homeostasis Sara E. Hanley1,And Katrina F
Preprints (www.preprints.org) | NOT PEER-REVIEWED | Posted: 6 November 2020 doi:10.20944/preprints202011.0241.v1 Sorting nexins in protein homeostasis Sara E. Hanley1,and Katrina F. Cooper2* 1Department of Molecular Biology, Graduate School of Biomedical Sciences, Rowan University, Stratford, NJ, 08084, USA 1 [email protected] 2 [email protected] * [email protected] Tel: +1 (856)-566-2887 1Department of Molecular Biology, Graduate School of Biomedical Sciences, Rowan University, Stratford, NJ, 08084, USA Abstract: Sorting nexins (SNXs) are a highly conserved membrane-associated protein family that plays a role in regulating protein homeostasis. This family of proteins is unified by their characteristic phox (PX) phosphoinositides binding domain. Along with binding to membranes, this family of SNXs also comprises a diverse array of protein-protein interaction motifs that are required for cellular sorting and protein trafficking. SNXs play a role in maintaining the integrity of the proteome which is essential for regulating multiple fundamental processes such as cell cycle progression, transcription, metabolism, and stress response. To tightly regulate these processes proteins must be expressed and degraded in the correct location and at the correct time. The cell employs several proteolysis mechanisms to ensure that proteins are selectively degraded at the appropriate spatiotemporal conditions. SNXs play a role in ubiquitin-mediated protein homeostasis at multiple levels including cargo localization, recycling, degradation, and function. In this review, we will discuss the role of SNXs in three different protein homeostasis systems: endocytosis lysosomal, the ubiquitin-proteasomal, and the autophagy-lysosomal system. The highly conserved nature of this protein family by beginning with the early research on SNXs and protein trafficking in yeast and lead into their important roles in mammalian systems. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Differential Expression of Hydroxyurea Transporters in Normal and Polycythemia Vera Hematopoietic Stem and Progenitor Cell Subpopulations
Zurich Open Repository and Archive University of Zurich Main Library Strickhofstrasse 39 CH-8057 Zurich www.zora.uzh.ch Year: 2021 Differential expression of hydroxyurea transporters in normal and polycythemia vera hematopoietic stem and progenitor cell subpopulations Tan, Ge ; Meier-Abt, Fabienne Abstract: Polycythemia vera (PV) is a myeloproliferative neoplasm marked by hyperproliferation of the myeloid lineages and the presence of an activating JAK2 mutation. Hydroxyurea (HU) is a standard treat- ment for high-risk patients with PV. Because disease-driving mechanisms are thought to arise in PV stem cells, effective treatments should target primarily the stem cell compartment. We tested for theantipro- liferative effect of patient treatment with HU in fluorescence-activated cell sorting-isolated hematopoietic stem/multipotent progenitor cells (HSC/MPPs) and more committed erythroid progenitors (common myeloid/megakaryocyte-erythrocyte progenitors [CMP/MEPs]) in PV using RNA-sequencing and gene set enrichment analysis. HU treatment led to significant downregulation of gene sets associated with cell proliferation in PV HSCs/MPPs, but not in PV CMP/MEPs. To explore the mechanism underlying this finding, we assessed for expression of solute carrier membrane transporters, which mediate trans- membrane movement of drugs such as HU into target cells. The active HU uptake transporter OCTN1 was upregulated in HSC/MPPs compared with CMP/MEPs of untreated patients with PV, and the HU diffusion facilitator urea transporter B (UTB) was downregulated in HSC/MPPs compared withCM- P/MEPs in all patient and control groups tested. These findings indicate a higher accumulation ofHU within PV HSC/MPPs compared with PV CMP/MEPs and provide an explanation for the differential effects of HU in HSC/MPPs and CMP/MEPs of patients with PV. -

Ingenuity Pathway Analysis of Differentially Expressed Genes Involved in Signaling Pathways and Molecular Networks in Rhoe Gene‑Edited Cardiomyocytes
INTERNATIONAL JOURNAL OF MOleCular meDICine 46: 1225-1238, 2020 Ingenuity pathway analysis of differentially expressed genes involved in signaling pathways and molecular networks in RhoE gene‑edited cardiomyocytes ZHONGMING SHAO1*, KEKE WANG1*, SHUYA ZHANG2, JIANLING YUAN1, XIAOMING LIAO1, CAIXIA WU1, YUAN ZOU1, YANPING HA1, ZHIHUA SHEN1, JUNLI GUO2 and WEI JIE1,2 1Department of Pathology, School of Basic Medicine Sciences, Guangdong Medical University, Zhanjiang, Guangdong 524023; 2Hainan Provincial Key Laboratory for Tropical Cardiovascular Diseases Research and Key Laboratory of Emergency and Trauma of Ministry of Education, Institute of Cardiovascular Research of The First Affiliated Hospital, Hainan Medical University, Haikou, Hainan 571199, P.R. China Received January 7, 2020; Accepted May 20, 2020 DOI: 10.3892/ijmm.2020.4661 Abstract. RhoE/Rnd3 is an atypical member of the Rho super- injury and abnormalities, cell‑to‑cell signaling and interaction, family of proteins, However, the global biological function and molecular transport. In addition, 885 upstream regulators profile of this protein remains unsolved. In the present study, a were enriched, including 59 molecules that were predicated RhoE‑knockout H9C2 cardiomyocyte cell line was established to be strongly activated (Z‑score >2) and 60 molecules that using CRISPR/Cas9 technology, following which differentially were predicated to be significantly inhibited (Z‑scores <‑2). In expressed genes (DEGs) between the knockout and wild‑type particular, 33 regulatory effects and 25 networks were revealed cell lines were screened using whole genome expression gene to be associated with the DEGs. Among them, the most signifi- chips. A total of 829 DEGs, including 417 upregulated and cant regulatory effects were ‘adhesion of endothelial cells’ and 412 downregulated, were identified using the threshold of ‘recruitment of myeloid cells’ and the top network was ‘neuro- fold changes ≥1.2 and P<0.05. -
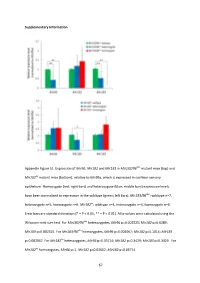
And Mir183 in Mir183/96 Dko Mutant Mice (Top) And
Supplementary Information Appendix Figure S1. Expression of Mir96 , Mir182 and Mir183 in Mir183/96 dko mutant mice (top) and Mir182 ko mutant mice (bottom), relative to Mir99a , which is expressed in cochlear sensory epithelium. Homozygote (red; right bars) and heterozygote (blue; middle bars) expression levels have been normalised to expression in the wildtype (green; left bars). Mir183/96 dko : wildtype n=7, heterozygote n=5, homozygote n=6. Mir182 ko : wildtype n=4, heterozygote n=4, homozygote n=4. Error bars are standard deviation (* = P < 0.05, ** = P < 0.01). All p-values were calculated using the Wilcoxon rank sum test. For Mir183/96 dko heterozygotes, Mir96 p=0.002525; Mir182 p=0.6389; Mir183 p=0.002525. For Mir183/96 dko homozygotes, Mir96 p=0.002067; Mir182 p=0.1014; Mir183 p=0.002067. For Mir182 ko heterozygotes, Mir96 p=0.05714; Mir182 p=0.3429; Mir183 p=0.3429. For Mir182 ko homozygotes, Mir96 p=1; Mir182 p=0.02652; Mir183 p=0.05714. 67 68 Appendix Figure S2. Individual ABR thresholds of wildtype, heterozygous and homozygous Mir183/96 dko mice at all ages tested. Number of mice of each genotype tested at each age is shown on the threshold plot. 69 70 Appendix Figure S3. Individual ABR thresholds of wildtype, heterozygous and homozygous Mir182 ko mice at all ages tested. Number of mice of each genotype tested at each age is shown on the threshold plot. 71 Appendix Figure S4. Mean ABR waveforms at 12kHz, shown at 20dB (top) and 50dB (bottom) above threshold (sensation level, SL) ± standard deviation, at four weeks old. -

Loss of Fam60a, a Sin3a Subunit, Results in Embryonic Lethality and Is Associated with Aberrant Methylation at a Subset of Gene
RESEARCH ARTICLE Loss of Fam60a, a Sin3a subunit, results in embryonic lethality and is associated with aberrant methylation at a subset of gene promoters Ryo Nabeshima1,2, Osamu Nishimura3,4, Takako Maeda1, Natsumi Shimizu2, Takahiro Ide2, Kenta Yashiro1†, Yasuo Sakai1, Chikara Meno1, Mitsutaka Kadota3,4, Hidetaka Shiratori1†, Shigehiro Kuraku3,4*, Hiroshi Hamada1,2* 1Developmental Genetics Group, Graduate School of Frontier Biosciences, Osaka University, Suita, Japan; 2Laboratory for Organismal Patterning, RIKEN Center for Developmental Biology, Kobe, Japan; 3Phyloinformatics Unit, RIKEN Center for Life Science Technologies, Kobe, Japan; 4Laboratory for Phyloinformatics, RIKEN Center for Biosystems Dynamics Research, Kobe, Japan Abstract We have examined the role of Fam60a, a gene highly expressed in embryonic stem cells, in mouse development. Fam60a interacts with components of the Sin3a-Hdac transcriptional corepressor complex, and most Fam60a–/– embryos manifest hypoplasia of visceral organs and die in utero. Fam60a is recruited to the promoter regions of a subset of genes, with the expression of these genes being either up- or down-regulated in Fam60a–/– embryos. The DNA methylation level of the Fam60a target gene Adhfe1 is maintained at embryonic day (E) 7.5 but markedly reduced at –/– *For correspondence: E9.5 in Fam60a embryos, suggesting that DNA demethylation is enhanced in the mutant. [email protected] (SK); Examination of genome-wide DNA methylation identified several differentially methylated regions, [email protected] (HH) which were preferentially hypomethylated, in Fam60a–/– embryos. Our data suggest that Fam60a is †These authors contributed required for proper embryogenesis, at least in part as a result of its regulation of DNA methylation equally to this work at specific gene promoters. -

Serum Albumin OS=Homo Sapiens
Protein Name Cluster of Glial fibrillary acidic protein OS=Homo sapiens GN=GFAP PE=1 SV=1 (P14136) Serum albumin OS=Homo sapiens GN=ALB PE=1 SV=2 Cluster of Isoform 3 of Plectin OS=Homo sapiens GN=PLEC (Q15149-3) Cluster of Hemoglobin subunit beta OS=Homo sapiens GN=HBB PE=1 SV=2 (P68871) Vimentin OS=Homo sapiens GN=VIM PE=1 SV=4 Cluster of Tubulin beta-3 chain OS=Homo sapiens GN=TUBB3 PE=1 SV=2 (Q13509) Cluster of Actin, cytoplasmic 1 OS=Homo sapiens GN=ACTB PE=1 SV=1 (P60709) Cluster of Tubulin alpha-1B chain OS=Homo sapiens GN=TUBA1B PE=1 SV=1 (P68363) Cluster of Isoform 2 of Spectrin alpha chain, non-erythrocytic 1 OS=Homo sapiens GN=SPTAN1 (Q13813-2) Hemoglobin subunit alpha OS=Homo sapiens GN=HBA1 PE=1 SV=2 Cluster of Spectrin beta chain, non-erythrocytic 1 OS=Homo sapiens GN=SPTBN1 PE=1 SV=2 (Q01082) Cluster of Pyruvate kinase isozymes M1/M2 OS=Homo sapiens GN=PKM PE=1 SV=4 (P14618) Glyceraldehyde-3-phosphate dehydrogenase OS=Homo sapiens GN=GAPDH PE=1 SV=3 Clathrin heavy chain 1 OS=Homo sapiens GN=CLTC PE=1 SV=5 Filamin-A OS=Homo sapiens GN=FLNA PE=1 SV=4 Cytoplasmic dynein 1 heavy chain 1 OS=Homo sapiens GN=DYNC1H1 PE=1 SV=5 Cluster of ATPase, Na+/K+ transporting, alpha 2 (+) polypeptide OS=Homo sapiens GN=ATP1A2 PE=3 SV=1 (B1AKY9) Fibrinogen beta chain OS=Homo sapiens GN=FGB PE=1 SV=2 Fibrinogen alpha chain OS=Homo sapiens GN=FGA PE=1 SV=2 Dihydropyrimidinase-related protein 2 OS=Homo sapiens GN=DPYSL2 PE=1 SV=1 Cluster of Alpha-actinin-1 OS=Homo sapiens GN=ACTN1 PE=1 SV=2 (P12814) 60 kDa heat shock protein, mitochondrial OS=Homo -

Decreased Function of Survival Motor Neuron Protein Impairs Endocytic
Decreased function of survival motor neuron protein PNAS PLUS impairs endocytic pathways Maria Dimitriadia,b, Aaron Derdowskic,1, Geetika Kallooa,1, Melissa S. Maginnisc,d, Patrick O’Herna, Bryn Bliskaa, Altar Sorkaça, Ken C. Q. Nguyene, Steven J. Cooke, George Poulogiannisf, Walter J. Atwoodc, David H. Halle, and Anne C. Harta,2 aDepartment of Neuroscience, Brown University, Providence, RI 02912; bDepartment of Biological and Environmental Sciences, University of Hertfordshire, Hatfield AL10 9AB, United Kingdom; cDepartment of Molecular Biology, Cell Biology, and Biochemistry, Brown University, Providence, RI 02912; dDepartment of Molecular and Biomedical Sciences, University of Maine, Orono, ME 04469; eDominick P. Purpura Department of Neuroscience, Albert Einstein College of Medicine, Bronx, NY 10461; and fChester Beatty Labs, The Institute of Cancer Research, London SW3 6JB, United Kingdom Edited by H. Robert Horvitz, Howard Hughes Medical Institute, Cambridge, MA, and approved June 2, 2016 (received for review January 23, 2016) Spinal muscular atrophy (SMA) is caused by depletion of the ubiqui- pathways most sensitive to decreased SMN is essential to un- tously expressed survival motor neuron (SMN) protein, with 1 in 40 derstand how SMN depletion causes neuronal dysfunction/death Caucasians being heterozygous for a disease allele. SMN is critical for in SMA and to accelerate therapy development. the assembly of numerous ribonucleoprotein complexes, yet it is still One of the early events in SMA pathogenesis is the loss of unclear how reduced SMN levels affect motor neuron function. Here, neuromuscular junction (NMJ) function, evidenced by muscle we examined the impact of SMN depletion in Caenorhabditis elegans denervation, neurofilament accumulation, and delayed neuro- and found that decreased function of the SMN ortholog SMN-1 per- muscular maturation (25–27). -

Low Affinity Uniporter Carrier Proteins Can Increase Net Substrate Uptake
www.nature.com/scientificreports OPEN Low afnity uniporter carrier proteins can increase net substrate uptake rate by reducing efux Received: 10 November 2017 Evert Bosdriesz 1,3, Meike T. Wortel 1,4, Jurgen R. Haanstra 1, Marijke J. Wagner1, Accepted: 9 March 2018 Pilar de la Torre Cortés2 & Bas Teusink 1 Published: xx xx xxxx Many organisms have several similar transporters with diferent afnities for the same substrate. Typically, high-afnity transporters are expressed when substrate is scarce and low-afnity ones when it is abundant. The beneft of using low instead of high-afnity transporters remains unclear, especially when additional nutrient sensors are present. Here, we investigate two hypotheses. It was previously hypothesized that there is a trade-of between the afnity and the catalytic efciency of transporters, and we fnd some but no defnitive support for it. Additionally, we propose that for uptake by facilitated difusion, at saturating substrate concentrations, lowering the afnity enhances the net uptake rate by reducing substrate efux. As a consequence, there exists an optimal, external-substrate- concentration dependent transporter afnity. A computational model of Saccharomyces cerevisiae glycolysis shows that using the low afnity HXT3 transporter instead of the high afnity HXT6 enhances the steady-state fux by 36%. We tried to test this hypothesis with yeast strains expressing a single glucose transporter modifed to have either a high or a low afnity. However, due to the intimate link between glucose perception and metabolism, direct experimental proof for this hypothesis remained inconclusive. Still, our theoretical results provide a novel reason for the presence of low-afnity transport systems. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase